Received: August 25, 2014
Accepted: December 12, 2014
Mediterr J Hematol Infect Dis 2015, 7(1): e2015012, DOI 10.4084/MJHID.2015.012
This article is available on PDF format at:

Roula A. Farah1*, Jessy G. Horkos1, Youssef D. Bustros2, Hussein Z. Farhat3 and Oussama Abla4
1 Department of Pediatrics, Division of Hematology/Oncology, Saint George Hospital University Medical Center, Beirut-Lebanon.
2 Department of Surgery, Saint George Hospital University Medical Center, Beirut-Lebanon.
3 Department of Laboratory Medicine, University Medical Center Rizk Hospital, Beirut- Lebanon.
4 Department of Pediatrics, Division of Hematology/Oncology, Toronto Sick Children’s Hospital.
|
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Background:
Acute myeloid leukemia (AML) is a disease with marked heterogeneity.
Despite major improvement in outcome, it remains a life-threatening
malignancy. Demographic and clinical data on pediatric AML is lacking
among the Lebanese population. Purpose: We aimed to identify clinical, molecular and outcome data in children with AML in Lebanon. Methods:
A retrospective chart review of children with AML diagnosed in three
Lebanese hospitals during the past 8 years was conducted. Results:
From May 2002 through March 2010, we identified 24 children with AML in
Saint George Hospital University Medical Center, University Medical
Center Rizk Hospital, and Abou-Jaoude Hospital. Males and females were
equally represented; median age at diagnosis was 9 years (range 1-24)
and median WBC at diagnosis was 31 × 109/L (range: 2.1-376 × 109/L).
Twenty five percent of patients (6 out of 24) had acute promyelocytic
leukemia (APL). Karyotype was normal in 33% of patients; t(8;21), inv
(16), t(8;9), t(7;11), t(9;11), complex chromosomal abnormality,
monosomy 7 and trisomy 8 were the most common cytogenetic abnormalities
encountered. Patients were treated on different European and North
American protocols. Twelve patients (50%) achieved morphologic CR after
cycle 1, 6 of them (50%) had bone marrow relapse within 11 months from
diagnosis. Nine patients underwent allogeneic stem cell transplant, and
3 of them are alive at 5 years post-transplant. Early death rate was
16.6% of patients, mainly those with APL and a presenting WBC > 10 ×
109/L. Fifty per cent of APL patients
had an early death due to DIC despite starting ATRA therapy. Overall,
median survival for AML patients who died from disease progression was
25.8 months (range: 1-60 months). Overall disease-free survival was
30.4%. Patients < 10 years of age had a 50% survival rate compared
to 0% in patients > 10 years. Conclusions:
Our report highlights the needs in Lebanon for better supportive care
of children with APL, including faster ATRA administration and,
aggressive transfusions, easy access to stem cell transplant for
high-risk AML patients and the need for a national homogenous treatment
strategy for children with AML. |
Introduction
Acute myeloid leukemia (AML) is composed of a group of diseases with
marked morphological and cytogenetic heterogeneity and accounts for
approximately 20% of childhood and adolescent acute leukemias.[1]
The majority of children with AML can achieve complete remission (CR)
when treated with conventional chemotherapy. However, despite
significant achievements in the treatment of AML, long-term survival
remains inferior to childhood acute lymphoblastic leukemia, even if, in
high-income countries, intensive therapy in conjunction with adequate
supportive care has increased survival rates to ∼ 70%, with event-free
survival rates (EFS) of ≥ 50%.[2]
Response to
therapy combined with genetic and molecular features are the most
important predictors of clinical outcome and are currently used for
risk stratification in most clinical trials.[3] There
are no published epidemiologic or clinical data regarding childhood AML
in Lebanon. In the present study we aimed to identify clinical,
morphological, molecular and outcome data for childhood and adolescent
AML in three Lebanese hospitals.
Methods
Results
Twenty-four patients were diagnosed with AML from May 2002 until March 2010 among the 3 different centers versus thirty nine patients with ALL. Presenting symptoms varied between fever, adenopathy, headache, malaise, bruising, abdominal pain, weight loss, decreased appetite and chloromas. Of the 24 patients, 12 were females (50%) and median age at presentation was 9 years. Seventeen patients (70.8%) were younger than 10 years at diagnosis. The median initial white blood cell count was 31x109/L (range: 2.1-376x109/L). All patients had low hemoglobin and platelet count at diagnosis, with a mean of 9.2 g/dl (range: 4.6-13.3 g/dl) and 46x109/L (range: 10-164x109/L), respectively. Three patients had secondary AML; one was previously treated for Burkitt lymphoma and 2 had Fanconi anemia and were siblings. Regarding the FAB subtypes, M3 was the most frequent (6 of 24; 25%) followed by M4 (5), M1 (4), M0 (2), M2 (2), M6 (2), M7 (1) and 2 patients were not classified. The M0, M1, and M2 subtypes tended to be more common in children > 10 years while the other subtypes were more frequent in those <10 years of age.[4] M1 was the most frequently observed subtype in five patients with normal karyotype (Table 1). Cytogenetic abnormalities were detected in 16 patients, and normal karyotype was detected in the others. Cytogenetic abnormalities included t(15;17) in 6 children, t(8;21) in 2, inv(16) in 2, t(8;9) in 1, t(7;11) in 1, t(9;11) in 1, complex chromosomal abnormality in 1, monosomy 7 in 1 and trisomy 8 in one child with Down syndrome (Figure 1). FLT3 gene mutations were detected in 3 patients; one had internal tandem duplication (ITD). However, molecular studies were not performed in all patients, and the real incidence of FLT3 mutations, as well as other mutations (such as NPM1 or CEBPA), could not be documented.
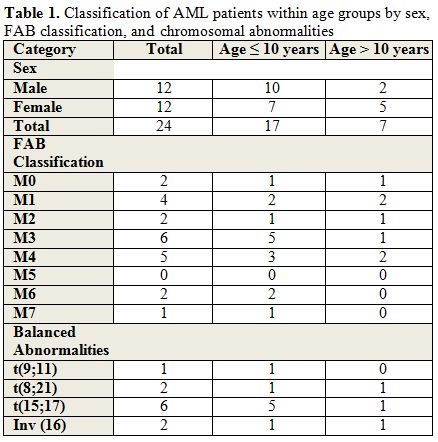 |
Table 1. Classification of AML patients within age groups by sex, FAB classification, and chromosomal abnormalities. |
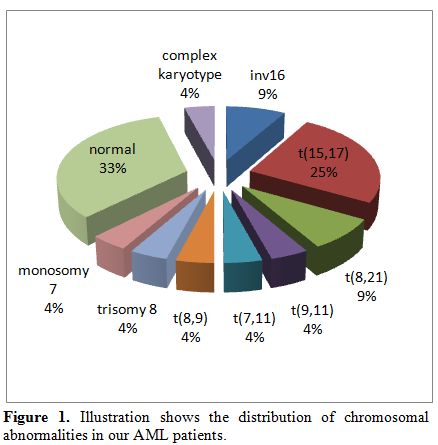 |
Figure 1. Illustration shows the distribution of chromosomal abnormalities in our AML patients. |
All patients were treated heterogenously (Table 2)
according to either the Berlin-Frankfurt-Munster (BFM)-98 protocol, St
Jude AML 97 protocol, the North American CALGB9710/Pediatric Oncology
Group (POG)-9421/Children’s Oncology Group (COG)-AAML0531 studies, the
French FLA protocol, Associazione Italiana di Ematologia Oncologia
Pediatrica (AIEOP)-AML 2002/01 or the Scandinavian NOPHO-AML protocols.
Twelve patients (50%) achieved morphologic CR (no minimal residual
disease testing was performed), 6 of them (50%) had bone marrow relapse
within eleven months from diagnosis. Hematopoietic stem cell transplant
(HSCT) was indicated in 15 patients, but only nine underwent the
procedure due to economical reasons or lack of a matched donor. Four
out of nine had matched related, and 5 had matched unrelated HSCT.
Three out of 9 children who underwent HSCT are currently alive at 5
years post-HSCT.
Children with AML < 10 years of age at
diagnosis had a 50% survival rate compared to 0% in children > 10
years resulting in worsening prognosis with rising age at presentation.[3]
The p value was 0.05 using Fisher exact test. There was no predilection
of gender that affected survival, nor a level of leukocytosis that
worsened the prognosis. Among the 15 patients who died, 8 had WBC <
31×109/L, and 7 had WBC > 31×109/L.
Three
out of six children (50%) with acute promyelocytic leukemia (APL) had
an early death; two died upon initiating of induction due to DIC and
CNS bleed, despite a quick diagnosis and early start of all-trans
retinoic acid (ATRA), while one died 2 days after diagnosis and before
starting any treatment. All 3 patients had an initial WBC > 109/L which is known to be a high-risk feature in APL.
Median
survival for patients who died from disease progression was 25.8
months. Overall disease-free survival was 30.4% at 5 years from
diagnosis.
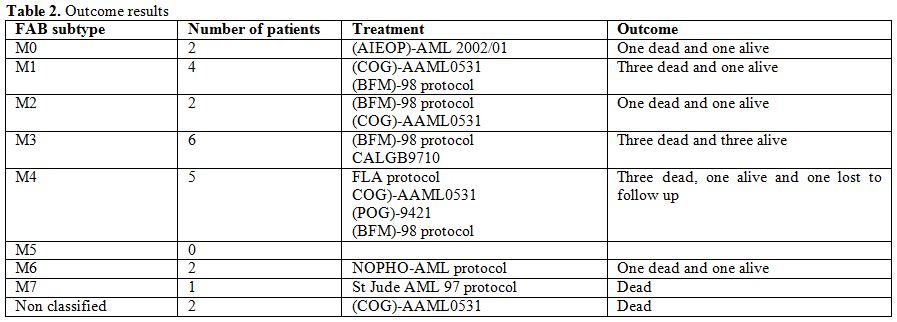 |
Table 2. Outcome results. |
Discussion
Acknowledgments
We thank Dr. Hanady Samaha, Dr. Laila Zahed, Dr. Noha Hakimeh, Dr. Mireille Zwein and Dr. Rami Mahfouz for their support with the diagnostic procedures. We also thank Dr Jihad Irani from Balamand University for his help with the statistical analysis.
References

[TOP]