Kwame Ofori Adjepong1, Folashade Otegbeye2 and Yaw Amoateng Adjepong3.
1 Warren Alpert Medical School, Brown University, United States.
2 Case Western Reserve University, University Hospitals Cleveland Medical Center, United States.
3 Yale University School of Medicine, Yale New Haven Health, Bridgeport Hospital, United States.
Corresponding
author: Yaw Amoateng Adjepong, MD MPH Ph.D. FACP. Assistant Clinical
Professor, Yale University School of Medicine. Senior Attending, Yale
New Haven Health, Bridgeport Hospital. 267 Grant Street, Bridgeport, CT
06610, USA. Tel: 203.374.4716, Fax: 203.384.3949. E-mail:
pyamoa@bpthosp.org
Published: May 1, 2018
Received: December 4, 2017
Accepted: April 19, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018032 DOI
10.4084/MJHID.2018.032
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Over
30 million people worldwide have sickle cell disease (SCD).
Emergent and non-emergent surgical procedures in SCD have been
associated with relatively increased risks of peri-operative mortality,
vaso-occlusive (painful) crisis, acute chest syndrome, post-operative
infections, congestive heart failure, cerebrovascular accident and
acute kidney injury. Pre-operative assessment must include a
careful review of the patient's known crisis triggers, baseline
hematologic profile, usual transfusion requirements, pre-existing organ
dysfunction and opioid use. Use of preoperative blood transfusions
should be selective and decisions individualized based on the baseline
hemoglobin, surgical procedure and anticipated volume of blood loss.
Intra- and post-operative management should focus on minimizing
hypoxia, hypothermia, acidosis, and intravascular volume depletion.
Pre- and post-operative incentive spirometry use should be encouraged.
|
Introduction
Over
30 million people worldwide, from 163 countries, have sickle cell
disease (SCD), with the highest concentration of the disease among
persons of African, Middle Eastern, and Central Indian ancestry.[1-3]
SCD patients undergo emergent and elective surgical procedures for SCD
complications and for surgical indications common to the general
population. Table 1 lists the
most common surgical procedures carried out in SCD patients. Surgeries
in the SCD population are associated with higher peri-operative
complication rates. We briefly review the epidemiology and
pathophysiology of SCD and discuss the major issues in the
peri-operative management of SCD patients.
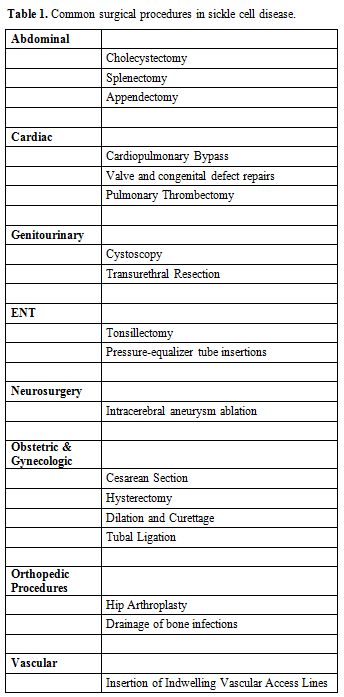 |
Table 1. Common surgical procedures in sickle cell disease. |
Ideally,
peri-operative care of the SCD patient must be a collaborative effort
between the surgeon, anesthetist, recovery room staff, primary care
physician, and a consulting hematologist experienced in the management
of SCD. Unfortunately, in many areas of the world, services from such
expert hematologists are not available. It is essential therefore for
all physicians who care for SCD patients to become familiar with the
perioperative management of SCD patients. This review is therefore
directed towards the global audience of generalist physicians,
surgeons, anesthetists, nurses, and intensivists involved in the
peri-operative management of SCD patients.
Genetics of Sickle Cell Disease
The normal adult hemoglobin, Hemoglobin A (HbA), is formed by two α and two β globin chains (α2β2),
clustered on chromosomes 16 and 11. The sickle hemoglobin mutation (Hb
S) results from a single amino acid substitution of valine for glutamic
acid in the 6th position of the β globin chain.[4] The sickle cell gene evolved independently in sub-Saharan Africa, the Arabian Peninsula and Central India.[2]
Several other hemoglobin gene variants have emerged in different
populations from spontaneous mutations. These are either structural
variants resulting from changes in the amino acid sequence or
thalassemias that lower or abolish globin chain production. Homozygous
inheritance of the sickle cell gene (Hb SS) or co-inheritance of the
sickle cell gene with another mutated hemoglobin gene variant results
in SCD.[1-7] The compound heterozygotes include a
combination of HbS with hemoglobin gene variants such as Hemoglobin C
(Hb SC), Hemoglobin D (Hb SD), Hemoglobin E (Hb SE), Hemoglobin O Arab
(Hb SO Arab) or with beta thalassemia (Hb Sβa thal). A combination of
HbS with the normal hemoglobin A results in the carrier state (Sickle
Cell trait), Hb AS, which is not considered sickle cell disease.[2] Globally the homozygous Hb SS, also called Sickle Cell Anemia, is the most common sickle cell genotype.[1,3-6]
The various sickle cell disease states differ in the percent HbS concentration (Table 2).[8]
This results in considerable heterogeneity in the phenotypic
manifestations of sickle cell disease, including the baseline
hemoglobin level. Overall, the most severe manifestations are seen in
the homozygous Hb SS and Hb Sβ0 thal genotypes.
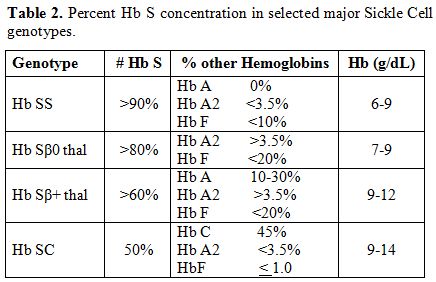 |
Table 2. Percent Hb S concentration in selected major Sickle Cell genotypes. |
Pathophysiology
The
hallmark of sickle cell disease is recurrent vaso-occlusion. It is now
apparent that complex and dynamic mechanisms underlie the
vaso-occlusive process, of which Hb S polymerization plays an essential
role.[9-15] Hemoglobin deoxygenation in SCD leads to
sickling of the RBCs as a result of Hb S polymerization. The process of
polymerization is triggered and/or enhanced by hypoxia, vascular
stasis, infection, inflammation, increased blood viscosity,
vasoconstriction, dehydration, hypotension, stress, cold temperature,
acidosis, and decreased flow.[9,10] There is RBC
clumping, endothelial damage, and inflammatory response with release of
mediators that up-regulate cell adhesion molecules on the endothelial
cells. These lead to adhesion of sickled cells to the vascular
endothelium, fibrin deposition and microvascular occlusion.
Neutrophils, monocytes, invariant natural killer T (iNKT) cells, other
leukocytes, and platelets are activated and participate in the vascular
occlusion. The vascular occlusion delays flow, further enhancing
deoxygenation of the affected red blood cells, leading to worsening
polymerization. The resulting ischemia creates a feedback loop of
deteriorating endothelial activation. These changes occur in multiple
vascular beds resulting in multisystem involvement. The half-life of
the sickled red cells is markedly reduced from the normal 120 days to
10 to 12 days, resulting in a chronic hemolytic anemia, with its
attendant hyper-proliferative bone marrow and hyperdynamic circulation,
even at baseline.[4,16,17] Surgical Complications
Historically,
surgical procedures in sickle cell patients have been associated with
relatively increased risks of peri-operative mortality, vaso-occlusive
(painful) crisis, acute chest syndrome, post-operative infections, and
congestive heart failure.[18-29] Careful pre-operative assessment and judicious peri-operative management are critical in mitigating these risks.
Pre-Operative Assessment and Interventions
The
goals of pre-operative assessment are to ensure that the patient is
medically optimized for the intended surgical procedure, estimate the
risk of peri-operative complications, and plan for the optimal
management of anticipated peri-operative complications. Sickle cell
disease must be appreciated as a multisystem disease that affects
almost all organs.[30]Currently, none of the generally available surgical risk calculators have been validated for patients with sickle cell disease.[31-33]
Risk estimates obtained using these surgical risk calculators, while
helpful, must be presumed to underestimate the overall peri-operative
risk in SCD patients.During
the pre-operative assessment one must ascertain the sickle cell
genotype, frequency of crisis and the date of patient's last crisis,
average length of hospital stay during painful crisis, known triggers
for crisis, baseline level of activity, baseline opioid use,
steady-state hemoglobin and hematocrit, reticulocyte count, and WBC
count, as well as history of blood transfusions. A sample pre-operative
data abstraction form is attached as Supplementary Appendix 1. The
severity of pre-existing cardiac and pulmonary complications of SCD
need to be ascertained in the pre-operative assessment. Table 3
lists selected known cardiac and pulmonary complications in SCD
patients. Routine pre-operative cardiac echography in all SCD
undergoing general anesthesia is unnecessary and will likely not impact
the peri-operative management in most patients. Nonetheless, given the
relatively large amounts of fluids administered to SCD patients,
cardiac echo may be useful to ascertain the extent of cardiac
dysfunction in patients with prior history of heart failure, poor
functional status or with dyspnea at baseline. In resource-poor
countries (where obtaining cardiac echography may not be readily
accessible), patients who develop breathlessness, fatigue or
palpitations and have to stop when walking at their own pace on a level
ground or when climbing a flight of stairs must be presumed to have
significant cardiac or pulmonary dysfunction. Inability to perform
these tasks will correspond to class III or higher on the New York
Heart Association heart failure classification,[35] or less than 4 metabolic equivalents functional activity level.[34] Extreme caution must be used in decisions about the volume of fluid administration in such patients.
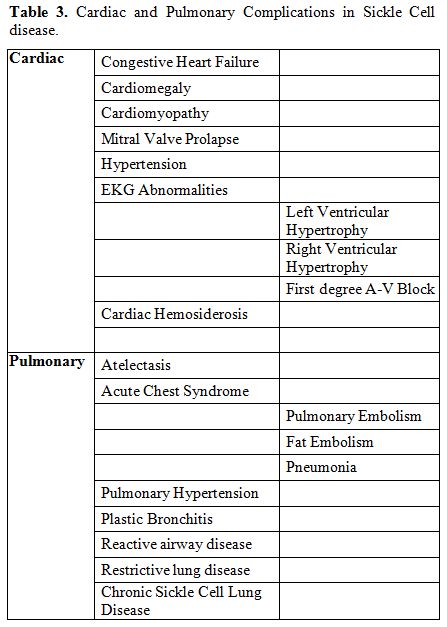 |
Table 3. Cardiac and Pulmonary Complications in Sickle Cell disease. |
Special steps should
be taken during the peri-operative process to avoid triggering a sickle
cell crisis. Common triggers of the acute crisis include anxiety,
emotional stress, infection, dehydration, acidosis, hypoxia, vascular
stasis and increased blood viscosity.[9,11]
Adequate counseling, including education of patient about the procedure
and awareness of patient’s special considerations, can significantly
assuage the emotional stress and anxiety about the surgical procedure.
If needed, anxiolytics can be used cautiously.Intracellular dehydration is a known trigger for Hb S polymerization.[9-11]
Whenever possible, prolonged pre-operative fasting must be avoided.
Patients should be encouraged to drink clear fluids up until 2-4 hours
before surgery. For patients undergoing moderate or major procedures,
intravenous hydration must be used. Supplement 2 lists the composition,
osmolarity and pH of commonly used solutions. Normal saline (9 g/dl of
sodium chloride contains 154 milliequivalents of sodium, pH of 5.5,
osmolarity of 308 milliosmoles per liter) is acidic and increases the
viscosity of the blood.[36] Hypotonic fluids, in theory, decrease RBC sickling and are preferred.[37-39]
Excessive fluid loading is associated with pulmonary edema and can
precipitate acute chest syndrome and thus needs to be avoided.[40,41] Exactly how much fluid should be given is unknown.[42]
Standard maintenance amounts may be used in most patients and the
intravenous infusion rate must be significantly reduced once the
patient resumes oral intake. Changes in daily weights and input/output
data may help guide the fluid management decisions.Hypoxia is the most important trigger of sickle cell crisis and needs to be avoided.[9-14,43] However, routine use of oxygen supplementation is not advisable as its potential harm far exceeds its benefits.[44-46]
To identify those in need of supplemental oxygen, oxygen monitoring in
the perioperative period must be considered mandatory in all patients.
Pulse oximetry does not correlate well with arterial oxygen tension in
some SCD patients.[47] It is therefore important that arterial blood gas confirmation is obtained in hypoxic patients. The
use of incentive spirometry has been shown to decrease the incidence of
atelectasis and acute chest syndrome in hospitalized patients.[40,48,49]
Accordingly, use of incentive spirometry before and after the procedure
needs to be strongly encouraged. Given the relatively high frequency of
acute chest syndrome following high risk (intracranial, cardiovascular,
and intrathoracic) procedures and the need to monitor arterial blood
gases during its management, establishing baseline blood gas values in
such patients is advised. Routine assessment of baseline pulmonary
function tests is not needed.[50,51]The
role of routine pre-operative blood transfusions, either simple RBC
transfusion or exchange transfusion, remains controversial.[27-29,52-68]
Theoretically, transfusion will reduce the percent HbS concentration
and improve tissue oxygen delivery. However, transfusion increases the
blood viscosity and thereby increases the risk of Hb polymerization.[55,56]
There is a net benefit of increased tissue oxygen delivery over
increased viscosity when the transfused hemoglobin level is kept at or
just below 10 g/dl.[55,56] However, no increase in
perioperative complications has been observed in centers that do not
routinely offer preoperative transfusion, or in countries with low
availability of blood for routine preoperative transfusion.[54]
Also, transfusion is associated with increased risks of
alloimmunization, iron overload and may be associated with increased
risk of infections.The
largest cohort study of surgery in SCD patients, the Cooperative Study
of Sickle Cell Disease, found beneficial effects of preoperative
transfusion in Hb SC patients for all surgical procedures.[27]
For Hb SS patients, peri-operative transfusion was associated with a
lower rate of SCD-related postoperative complications in patients
undergoing low-risk procedures (such as inguinal hernia repair,
myringotomy, dilatation and curettage, and surgeries on eyes, skin, and
nose). However, there was no association between transfusion and
sickle-related postoperative complications among patients who had
moderate risk procedures (throat, neck, spine, proximal extremities,
hip replacement, genitourinary system, and intra-abdominal).[27] Using
a target hemoglobin level of 10 g/dl, the Preoperative Transfusion in
Sickle Cell Disease study Group performed a multicenter, randomized
controlled trial and found conservative, simple RBC transfusion was
equally effective as exchange transfusion (maintaining hemoglobin at
10g/dl and HbS level of 30% or less) in preventing peri-operative
complications.[58] However, the study did not have a
comparable group without blood transfusion. Also, transfusion was
associated with increased rates of allo-immunization. Recently, the Transfusion Alternatives Preoperatively In Sickle Cell Disease (TAPS) study,[53] a multicenter randomized trial of 67 Hb SS and Hb Sβ0
thal patients, found a reduction in clinically important complications
in the transfused patients undergoing medium risk procedures (15% vs.
39%, p=0.02). In contrast, preoperative transfusion was associated with
a higher rate of post-operative complications in a matched prospective
study of 40 patients undergoing laparoscopic cholecystectomy for
cholelithiasis in Saudi Arabia (25% vs. 0%, p=0.007).[59]Tables 4 and 5 summarize the findings of published original studies on pre-operative transfusions.[27-29,53,57-68]
A systematic review and meta-analysis of the randomized and
observational studies found no difference in perioperative mortality,
vascular, or non-vascular perioperative complications between those
treated with preoperative transfusion versus no transfusion strategy.[52]
This review notwithstanding, the current consensus in the United States
is “to bring the hemoglobin level to 10 g/dl prior to undergoing a
surgical procedure involving general anesthesia” in patients with Hb SS
or Sβ0 thal.[56]
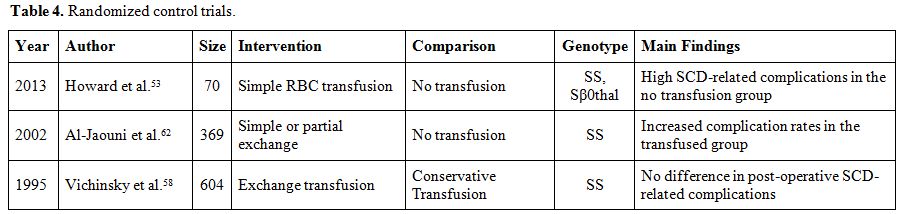 |
Table 4. Randomized control trials. |
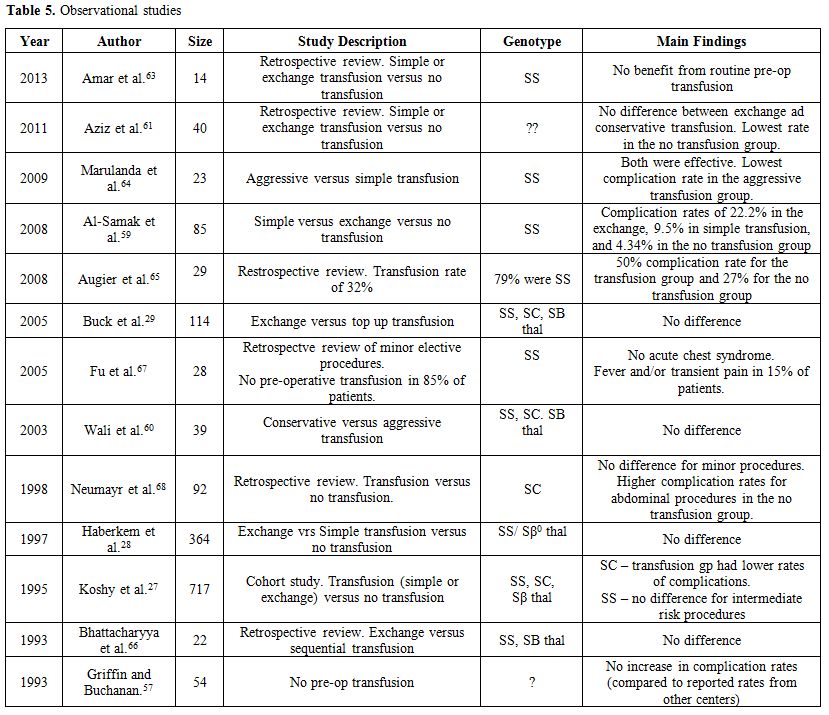 |
Table 5. Observational studies |
Based
on the current aggregate data, it is fair to advocate that transfusion
decisions need to be selective and individualized based on the type of
SCD, the baseline hemoglobin, the baseline cardiopulmonary reserve, and
the risk of the surgical procedure. If a decision to transfuse is made,
phenotypically matched blood must be used to minimize the risk of
alloimmunization. For those with hemoglobin levels less than 9g/dl,
simple RBC transfusion is equally efficacious compared to exchange
transfusion. For those with high baseline hemoglobin (above 9 g/dl),
perhaps exchange (or partial exchange) transfusion, rather than simple
transfusion, should be used to avoid raising the hemoglobin level above
10 g/dl.Cold weather and skin chilling are known precipitants of the crisis in some sickle cell patients.[9,10]
It has been hypothesized that hypothermia leads to exaggerated reflex
vasoconstriction, increased capillary transit time, red cell sludging,
and may lead to shunting of blood from the bone marrow.[15,69,70]
Accordingly, thermoregulation has been strongly recommended in the
perioperative care of SCD patients. Warm intravenous fluids are
advised. Many centers fearfully avoid hypothermia, even in cardiac
surgery, in sickle cell patients. To date, there are no known reports
of peri-operative hypothermia as a contributory cause of perioperative
vaso-occlusive crisis. In vitro, there is delayed RBC sickling and
slowing of polymerization with hypothermia. Indeed, hypothermic
cardiopulmonary bypass, including cold crystalloid cardioplegia and
systemic hypothermia, have been and continue to be successfully used in
cardiac surgery in sickle cell patients at one major center without any
significant adverse effects.[54,71]
This center’s protocol meticulously avoids hypoxia, acidosis,
hypotension, and dehydration in these patients. It has been suggested
that the level of anesthesia needed for cardiopulmonary bypass impairs
thermoregulatory vasoconstriction, the presumed mechanism of
hypothermia-induced sickling.[25,54]
Anesthetic Agents
In
general, anesthetic techniques used in SCD patients must minimize
exposure to hypoxemia, hypercapnia, acidosis, hypothermia, and
hypovolemia during surgery. Care with positioning is important to
minimize venous stasis. Respiratory depressants are avoided.
Intubations are usually performed after paralysis with a short-acting
agent. During induction, steps are taken to avoid breath holding,
laryngeal spasm and struggling. A variety of anesthetic agents have
been used successfully and the choice of a specific study, the use of
halothane for anesthesia was associated with the lowest risk of
perioperative atelectasis (25%) as compared with isoflurane (83%) or
enflurane (59%).[72] The choice of agent used had no effect on SCD-related morbidity.The
relative safety of general anesthesia compared to regional anesthesia
in SCD patients is unclear. SCD related complications were more
frequent in those who received regional anesthesia compared with those
who received general anesthesia in the Cooperative Study of Sickle Cell
Disease.[27] However, there was no adjustment for
potential confounding by the effect of obstetric procedures, for which
regional procedures were more often used. Similarly, regional
procedures were often used for sicker patients who were considered too
high a risk for general anesthesia. Other studies have failed to
confirm such an association between regional anesthesia and increased
complications.[73,74] Theoretically, in regional
anesthesia there is regional hypoperfusion, venous stasis, and lack of
control of ventilation. There is a redistribution of blood flow with
increase in capillary and venous oxygen tension in the blocked region,
and compensatory vasoconstriction in the non-blocked area with
resultant fall in oxygen.
Surgical Procedures
The
uses of laparoscopic procedures have significantly shortened hospital
stay. Among SCD patients, laparoscopic cholecystectomy and laparoscopic
splenectomy are preferred compared to open cholecystectomy or
splenectomy. In many centers, it has been associated with significant
reductions in post-operative complications.[75-77] In one center, however, laparoscopic surgery did not decrease the risk of developing ACS.[78]The
use of an arterial tourniquet in SCD patients is controversial. It is
dogma that application of arterial tourniquet creates ideal conditions
for sickling from the stasis, hypoxia, and acidosis distal to the
tourniquet. As such, SCD has long been considered a contraindication to
tourniquet use.[79] This dogma is now being questioned.[80-83]
Tourniquets have been used successfully in SCD patients with acceptable
or no complications, while paying “meticulous attention to preoperative
preparation and intraoperative management”.[83] It
has been postulated that the acute acidotic environment induced by the
tourniquet application alters endothelium-RBC membrane interactions,
promote systemic vasodilatation, and “alter a host of other biochemical
reactions” that on balance may not promote sickling.[83]
No randomized studies have been conducted. A review of the rather
limited published reports suggests that tourniquets may be used with
relative safety in most patients with sickle cell disease with proper
perioperative management.[84] Radiological Contrast
Contrast
imaging studies are often needed in cardiac and neurologic surgeries.
Hyperosmolar contrast media can induce RBC dehydration, polymerization
and sickling, with a resultant sickle crisis.[85] Isotonic media have no such deleterious effects.[86] Accordingly, it is recommended that low osmolar or isotonic contrast media be used in sickle cell disease patients.[86] Radiologic contrast should be avoided in SCD patients with renal failure.
Postoperative Care
Pain control.
An important issue in the perioperative management of sickle cell
disease patients is adequate pain control. Nonpharmacologic measures,
including music, relaxation, heat or ice packs, may be used as adjuncts
to pharmacologic pain management.[56] The patient's
self-report, in addition to the vital signs, needs to be incorporated
in adjusting pain medications. Many adult SCD patients in the US have
had multiple exposures to opioids, are often opioid-tolerant, and tend
to require large doses of opiates for adequate pain control.[56,87]
If known, patients’ opioid doses used for management of their painful
crises can serve as a guide to post-operative pain management. A
combination of long-acting opioids and a short-acting opioid for
breakthrough pain often provides adequate relief. Alternatively,
continuous administration of pain medications, through the use of
patient-controlled analgesia pumps, may be used. Morphine and
hydromorphone are the major opioid agonists used for severe pain
management in sickle cell patients in the post-operative period. These
drugs have no ceiling effect. However, they can cause severe sedation
and respiratory depression. Hence, doses should be discontinued or
skipped in patients with a respiratory rate less than 10 and in those
with severe sedation.Acute chest syndrome.
Sickle cell patients are at risk for acute chest syndrome in the
immediate post-operative period. Excessive administration of IV fluids,
as well as respiratory sedation from the use of opioid medications and
adjuvants, potentiate this risk.[40] Maintaining
adequate ventilation is the best preventive measure. Pre and
post-operative use of incentive spirometry is strongly advised. The
role of prophylactic CPAP in the immediate post-operative period has
yet to be evaluated. Fluid administration should not exceed one and
one-half (1.5) times the patient’s maintenance requirements.[40,56] Prompt
recognition of acute chest syndrome is important. By definition, acute
chest syndrome is the presence of a new pulmonary infiltrates with
chest pain, tachypnea, hypoxia, dyspnea, cough, fever, or leukocytosis.[30,40,56]
However, not all the cardinal signs and symptoms may be present
initially. The spectrum of presentation may range from mild, where
hypoxia is minimal, to severe acute respiratory distress. Management
consists of ensuring adequate ventilation, including the use of
mechanical ventilation in severe cases, oxygen administration,
bronchodilators (even in the absence of wheezing), antibiotics,
moderate use of analgesia, and judicious hydration.[56]
Simple blood transfusion or exchange transfusion in severe cases can
accelerate the resolution. The use of steroids, particularly in adult
patients, is controversial.[56] The use of nitric
oxide or other vasodilators (calcium channel blockers, prostacyclin),
and the nonionic surfactant poloxamer 188, is currently undergoing
clinical trials.[56]Deep vein thrombosis prophylaxis. Sickle cell disease is a hypercoagulable state.[88-90]
Current evidence suggests increased platelet and coagulation
activation, even at the patient’s basal state. SCD patients have low
circulating levels of anticoagulant proteins C and S, moderate
thrombocytosis, decreased platelet thrombospondin-1 content, and
increased levels of markers of platelet activation.[88-90]
Adequate deep vein thrombosis prophylaxis must be instituted after all
major surgeries until the patients are sufficiently ambulatory. Post-operative fever.
Post-operative fever is a common complication of many major surgical
procedures in the general population, with estimates ranging from 14%
to 91% depending on the type of procedure.[91,92]
Major traumatic surgeries are associated with higher risks of
postoperative fever. Highest fever rates are observed after major
orthopedic procedures.[91] Interleukin – 6 is an
important driver of this response. Fever tends to be non-infectious in
etiology if it occurs within the intra-operative period or in the first
48-hours. Other non-infectious causes include administration of blood
products, heparin, and other medications. Infections account for most
fevers occurring after the second postoperative day.[91,92] Reported rates of post-operative fever among SCD patients are comparable to rates for non-SCD patients.[27,93,94]
However, because of the higher rates of functional asplenia in SCD
patients, they are more susceptible to invasive bacterial infections
from encapsulated organisms such as Streptococcus pneumonia and
Hemophilus influenza. These infections can be overwhelming if therapy
is delayed. Fortunately, immunization with pneumococcal and Hemophilus
influenza vaccinations have significantly decreased these risks in many
countries. Nonetheless, the occurrence of post-operative fever in SCD
requires careful clinical and laboratory evaluation. The extent of
diagnostic work-up must be guided by the history and physical
examination findings. Fever occurring after 48 hours must be managed as
infectious in origin until proven otherwise. Common causes of infection
include urinary tract infections, pneumonia, intravascular
catheter-related infections, surgical site infections, and/or infected
prosthesis. Viral infections from transfused blood products are now
rare and tend to occur after 4 weeks.[92] Conclusions
In
conclusion, pre-operative assessment of SCD patients undergoing
emergent or non-emergent surgery must include a careful review of the
patient's known crisis triggers, baseline hematologic profile, standard
transfusion requirements, pre-existing organ dysfunction and opioid
use. Use of preoperative blood transfusions should be selective, and
decisions must be individualized based on the baseline hemoglobin,
surgical procedure and anticipated volume of blood loss. Intra- and
post-operative management should focus on minimizing hypoxia,
hypothermia, acidosis, and intravascular volume depletion. Excessive
administration of IV fluids, as well as respiratory sedation from the
use of opioid medications and adjuvants, potentiate the risk of acute
chest syndrome. Use of pre- and post-operative incentive spirometry
should be strongly encouraged. Arterial tourniquets and hypothermic
cardiopulmonary bypass have been safely used in SCD patients at some
centers.
Take Home Points
1.Surgical
procedures in SCD have been associated with relatively increased risks
of peri-operative mortality, vaso-occlusive (painful) crisis, acute
chest syndrome, post-operative infections, congestive heart failure and
acute kidney injury.
2.Use of preoperative blood transfusions should be selective.
3.Intra-
and post-operative management should focus on minimizing hypoxia,
hypothermia, acidosis, and intravascular volume depletion.
4.Pre- and post-operative use of incentive spirometry decreases the risk of acute chest syndrome.
5.Use
of arterial tourniquets and hypothermic cardiopulmonary bypass in SCD
patients, though controversial, have been safely utilized at some
centers.
References
- Modell B, Darlison MW, Moorthie S, Blencowe H,
Petrou M, Lawn J. Epidemiologic methods in community genetics and model
global database of congenital disorders. 2016 (in Press).

- Tsaras
G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications
associated with sickle cell trait: a brief narrative review. The
American Journal of Medicine, 2009; 122(6): 507-512. https://doi.org/10.1016/j.amjmed.2008.12.020 PMid:19393983
- Piel
FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of
sickle cell anaemia in children under five, 2010–2050: modelling based
on demographics, excess mortality, and interventions. PLoS Medicine.
2013 Jul 16;10(7):e1001484.
- Ashley-Koch
A, Yang Q, Olney R S. Sickle hemoglobin (Hb S) allele and sickle cell
disease: a HuGE review. American Journal of Epidemiology 2000; 151(9):
839-845. https://doi.org/10.1093/oxfordjournals.aje.a010288 PMid:10791557
- Steinberg
MH, Rodgers GP. Pathophysiology of sickle cell disease: role of
cellular and genetic modifiers. Seminars in Hematology 2001; 38 (4):
299-306. https://doi.org/10.1016/S0037-1963(01)90023-X
- Hassell
K L. Population estimates of sickle cell disease in the US. American
Journal of Preventive Medicine 2010; 38(4): S512-S521.
- Shafer
FE, Lorey F, Cunningham GC, et al. Newborn screening for sickle cell
disease: 4 years of experience from California's newborn screening
program. Journal of Pediatric Hematology/Oncology 1996; 18(1): 36-41. https://doi.org/10.1097/00043426-199602000-00007 PMid:8556368
- National
Heart, Lung, and Blood Institute. Evidence-based management of sickle
cell disease: expert panel report, 2014. Washington, DC: National
Institutes of Health. 2014.
- Noguchi CT, Schechter AN, Rodgers GP. Sickle cell disease pathophysiology. Baillière's Clinical Haematology 1993; 6(1): 57-91. https://doi.org/10.1016/S0950-3536(05)80066-6
- Kato
GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell
disease: Biology, pathophysiology, genetics, translational medicine,
and new research directions. American Journal of Hematology 2009;
84(9): 618-625. https://doi.org/10.1002/ajh.21475 PMid:19610078 PMCid:PMC3209715
- Manwani
D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology
and novel targeted therapies. Blood 2013; 122(24): 3892-3898. https://doi.org/10.1182/blood-2013-05-498311 PMid:24052549 PMCid:PMC3854110
- Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB life 2012; 64(1): 72-80. https://doi.org/10.1002/iub.584 PMid:22131167
- Kassim AA, DeBaun MR. Sickle cell disease, vasculopathy, and therapeutics. Annual Review of Medicine 2013; 64: 451-466. https://doi.org/10.1146/annurev-med-120611-143127 PMid:23190149
- Frenette
PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm.
Current Opinion in Hematology 2002; 9(2): 101-106. https://doi.org/10.1097/00062752-200203000-00003 PMid:11844991
- Kaul DK, Finnegan E, Barabino GA. Sickle red cell–endothelium interactions. Microcirculation 2009; 16(1): 97-111. https://doi.org/10.1080/10739680802279394 PMid:18720225 PMCid:PMC3059190
- McCurdy
PR, Sherman AS. Irreversibly sickled cells and red cell survival in
sickle cell anemia: a study with both DF32P and 51CR. The American
Journal of Medicine 1978; 64(2): 253-258. https://doi.org/10.1016/0002-9343(78)90053-0
- Eadie GS, Brown IW, Analytical review: red blood cell survival studies. Blood 1953; 8(12): 1110-1136. PMid:13105714
- Holzmann
L, Finn H, Lichtman HC, Harmel MH. Anesthesia in patients with sickle
cell disease: a review of 112 cases. Anesthesia & Analgesia 1969;
48(4): 566-572. https://doi.org/10.1213/00000539-196907000-00013
- Patterson
RH, Wilson H, Diggs LW. Sickle-Cell Anemia: a Surgical Problem. II.
Further Observation on the Surgical Implications of Sickle-Cell Anemia.
Surgery 1950; 28(2): 393-402. PMid:15442815
- Browne RA. Anaesthesia in patients with sickle-cell anaemia. British Journal of Anaesthesia 1965; 37(3): 181-188. https://doi.org/10.1093/bja/37.3.181 PMid:14277955
- Shapiro ND, Poe MF. Sickle-cell disease: an anesthesiological problem. Anesthesiology 1955; 16(5): 771. https://doi.org/10.1097/00000542-195509000-00017
- Oduro KA. Anaesthesia and sickle cell haemoglobin. British Journal of Anaesthesia 1973; 45(1):123. https://doi.org/10.1093/bja/45.1.123-a PMid:4696431
- Oduro KA, Searle JF. Anaesthesia in sickle-cell states: a plea for simplicity. British Medical Journal 1972; 4(5840): 596-8. https://doi.org/10.1136/bmj.4.5840.596
- Searle JF. Anaesthesia in sickle cell states. Anaesthesia 1973; 28(1): 48-58. https://doi.org/10.1111/j.1365-2044.1973.tb00284.x PMid:4568880
- Firth PG, Head CA. Sickle cell disease and anesthesia. Anesthesiology 2004;101: 766-786. https://doi.org/10.1097/00000542-200409000-00027
- Vichinsky
EP, Neumayr LD, Haberkern C, et al. The perioperative complication rate
of orthopedic surgery in sickle cell disease: report of the National
Sickle Cell Surgery Study Group. American Journal of Hematology 1999,
62(3), 129-138. https://doi.org/10.1002/(SICI)1096-8652(199911)62:3<129::AID-AJH1>3.0.CO;2-J
- Koshy
M, Weiner SJ, Miller ST, et al. Surgery and anesthesia in sickle cell
disease. Cooperative Study of Sickle Cell Diseases. Blood 1995; 86(10):
3676-3684. PMid:7579333
- Haberkern
CM, Neumayr LD, Orringer EP, et al. Cholecystectomy in sickle cell
anemia patients: perioperative outcome of 364 cases from the National
Preoperative Transfusion Study. Blood 1997; 89(5):1533-1542.
PMid:9057634
- Buck J, Davies SC. Surgery in sickle cell disease. Hematology/oncology clinics of North America 2005; 19(5): 897-902. https://doi.org/10.1016/j.hoc.2005.07.004 PMid:16214650
- Ballas
SK, Lieff S, Benjamin LJ, et al. Definitions of the phenotypic
manifestations of sickle cell disease. Am J Hematol. 2010
Jan;85(1):6-13 PMid:19902523 PMCid:PMC5046828
- Gupta
PK, Gupta H, Sundaram A, Kaushik M, Fang X, Miller WJ, Esterbrooks DJ,
Hunter CB, Pipinos II, Johanning JM, Lynch TG. Development and
validation of a risk calculator for prediction of cardiac risk after
surgery. Circulation. 2011 Jan 1: CIRCULATIONAHA-110.
- Bilimoria
KY, Liu Y, Paruch JL, Zhou L, Kmiecik TE, Ko CY, Cohen ME. Development
and evaluation of the universal ACS NSQIP surgical risk calculator: a
decision aid and informed consent tool for patients and surgeons.
Journal of the American College of Surgeons. 2013 Nov 1;217(5):833-42. https://doi.org/10.1016/j.jamcollsurg.2013.07.385 PMid:24055383 PMCid:PMC3805776
- Ford
MK, Beattie WS, Wijeysundera DN. Systematic review: prediction of
perioperative cardiac complications and mortality by the revised
cardiac risk index. Annals of Internal Medicine. 2010 Jan
5;152(1):26-35. https://doi.org/10.7326/0003-4819-152-1-201001050-00007 PMid:20048269
- Jette
M, Sidney K, Blümchen G. Metabolic equivalents (METS) in exercise
testing, exercise prescription, and evaluation of functional capacity.
Clinical Cardiology. 1990 Aug 1;13(8):555-65. https://doi.org/10.1002/clc.4960130809 PMid:2204507
- Kubo
SH, Schulman S, Starling RC, Jessup M, Wentworth D, Burkhoff D.
Development and validation of a patient questionnaire to determine New
York Heart Association classification. Journal of Cardiac Failure. 2004
Jun 1;10(3):228-35. https://doi.org/10.1016/j.cardfail.2003.10.005 PMid:15190533
- Coran
AG, Ballantine TV, Horwitz DL, Herman CM. The effect of crystalloid
resuscitation in hemorrhagic shock on acid-base balance: A comparison
between normal saline and Ringer's lactate solutions. Surgery 1971;,
69(6): 874-880. PMid:5578448
- Guy
RB, Gavrilis PK, Rothenberg SP. In vitro and in vivo effect of
hypotonic saline on the sickling phenomenon. The American Journal of
the Medical Sciences 1973; 266(4): 267-277. https://doi.org/10.1097/00000441-197310000-00005 PMid:4757805
- Carden
MA, Fay M, Sakurai Y, McFarland B, Blanche S, DiPrete C, Joiner CH,
Sulchek T, Lam WA. Normal saline is associated with increased sickle
red cell stiffness and prolonged transit times in a microfluidic model
of the capillary system. Microcirculation. 2017 Jul 1;24(5). https://doi.org/10.1111/micc.12353 PMid:28106307
- Carden
MA, Fay ME, Lu X, Mannino RG, Sakurai Y, Ciciliano JC, Hansen CE,
Chonat S, Joiner CH, Wood DK, Lam WA. Extracellular fluid tonicity
impacts sickle red blood cell deformability and adhesion. Blood. 2017
Dec 14;130(24):2654-63. https://doi.org/10.1182/blood-2017-04-780635 PMid:28978568
- Vichinsky
EP, Styles LA, Colangelo LH et al. Acute chest syndrome in sickle cell
disease: clinical presentation and course. Blood 1997; 89(5),
1787-1792. PMid:9057664
- Haynes
J, Allison RC. Pulmonary edema: complication in the management of
sickle cell pain crisis. The American Journal of Medicine 1986; 80(5):
833-840. https://doi.org/10.1016/0002-9343(86)90624-8
- Jones J, Quinn R. Fluid Replacement Strategies in Sickle Cell Disease. Proceedings of UCLA Healthcare. 2017;21.
- Kaul
DK, Fabry ME, Costantini F, Rubin EM, Nagel RL. In vivo demonstration
of red cell-endothelial interaction, sickling and altered microvascular
response to oxygen in the sickle transgenic mouse. Journal of Clinical
Investigation 1995; 96(6): 2845. https://doi.org/10.1172/JCI118355 PMid:8675655 PMCid:PMC185995
- Embury
SH, Garcia JF, Mohandas N, Pennathur-Das R, Clark MR. Effects of oxygen
inhalation on endogenous erythropoietin kinetics, erythropoiesis, and
properties of blood cells in sickle-cell anemia. New England Journal of
Medicine 1984; 311(5): 291-295. https://doi.org/10.1056/NEJM198408023110504 PMid:6738642
- Zipursky
A, Robieux IC, Brown EJ, et al. Oxygen therapy in sickle cell disease.
Journal of Pediatric Hematology/Oncology 1992; 14(3): 222-228. https://doi.org/10.1097/00043426-199208000-00007
- Reinhard
EH, Moore CV, Dubach R, Wade LJ. Depressant effects of high
concentrations of inspired oxygen on erythrocytogenesis. Observations
on patients with sickle cell anemia with a description of the observed
toxic manifestations of oxygen. Journal of Clinical Investigation 1944;
23(5): 682. https://doi.org/10.1172/JCI101539 PMid:16695150 PMCid:PMC435388
- Blaisdell
CJ, Goodman S, Clark K, Casella JF, Loughlin GM. Pulse oximetry is a
poor predictor of hypoxemia in stable children with sickle cell
disease. Archives of Pediatrics & Adolescent Medicine 2000; 154(9):
900-903. https://doi.org/10.1001/archpedi.154.9.900
- Bellet
PS, Kalinyak KA, Shukla R, Gelfand MJ, Rucknagel DL. Incentive
spirometry to prevent acute pulmonary complications in sickle cell
diseases. New England Journal of Medicine 1995; 333(11): 699-703. https://doi.org/10.1056/NEJM199509143331104 PMid:7637747
- Niss
O, Cole-Jenkins C, Davis B, Brooks T, Woolery K, Fetters T, Rollins J,
McGann PT, Kalinyak K. Prevention of Acute Chest Syndrome By
Implementing a Standardized Process to Improve Incentive Spirometry Use
in Hospitalized Patients with Sickle Cell Disease. Blood 2017; 130
(Suppl 1):132.
- Jacob
B, Amoateng-Adjepong Y, Rasakulasuriar S, Manthous CA, Haddad R.
Preoperative pulmonary function tests do not predict outcome after
coronary artery bypass. Connecticut Medicine 1997; 61(6): 327-332.
PMid:9238826
- Smetana G W. Preoperative pulmonary evaluation. New England Journal of Medicine 1999; 340(12): 937-944. https://doi.org/10.1056/NEJM199903253401207 PMid:10089188
- Alotaibi
GS, Alsaleh K, Bolster L, Sean McMurtry M, Wu C. Preoperative
transfusion in patients with sickle cell disease to prevent
perioperative complications: A systematic review and meta-analysis.
Hematology 2014; 19(8): 463-471. https://doi.org/10.1179/1607845414Y.0000000158 PMid:24611757
- Howard
J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives
Preoperatively in Sickle Cell Disease (TAPS) study: a randomised,
controlled, multicentre clinical trial.The Lancet 2013; 381(9870):
930-938. https://doi.org/10.1016/S0140-6736(12)61726-7
- Edwin
F, Aniteye E, Tettey M, et al. Hypothermic cardiopulmonary bypass
without exchange transfusion in sickle-cell patients: a matched-pair
analysis. Interactive cardiovascular and thoracic surgery 2014; 19(5):
771-776. https://doi.org/10.1093/icvts/ivu249 PMid:25080509
- Davies SC, Roberts-Harewood M. Blood transfusion in sickle cell disease. Blood Reviews 1997; 11(2): 57-71. https://doi.org/10.1016/S0268-960X(97)90012-6
- Yawn
B P, Buchanan GR, Afenyi-Annan AN, et al. (2014). Management of sickle
cell disease: summary of the 2014 evidence-based report by expert panel
members. JAMA 2014; 312(10):1033-1048. https://doi.org/10.1001/jama.2014.10517 PMid:25203083
- Griffin
TC, Buchanan GR. Elective surgery in children with sickle cell disease
without preoperative blood transfusion. Journal of Pediatric Surgery
1993; 28(5): 681-685. https://doi.org/10.1016/0022-3468(93)90031-F
- Vichinsky
EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and
aggressive transfusion regimens in the perioperative management of
sickle cell disease. The Preoperative Transfusion in Sickle Cell
Disease Study Group. N Engl J Med. 1995; 333(4):206-13. https://doi.org/10.1056/NEJM199507273330402 PMid:7791837
- Al-Samak
ZM, Al-Falaki MM, Pasha AA. Assessment of perioperative transfusion
therapy and complications in sickle cell disease patients undergoing
surgery. Middle East J Anesthesiol. 2008;19(5):983-95. PMid:18637600
- Wali
YA, al Okbi H, al Abri R. A comparison of two transfusion regimens in
the perioperative management of children with sickle cell disease
undergoing adenotonsillectomy. Pediatr Hematol Oncol. 2003; 20(1):7-13.
https://doi.org/10.1080/0880010390158487 PMid:12687748
- Aziz
AM, Meshikhes AW. Blood transfusion in patients with sickle cell
disease requiring laparoscopic cholecystectomy. JSLS: Journal of the
Society of Laparoendoscopic Surgeons. 2011 Oct;15(4):480. https://doi.org/10.4293/108680811X13176785203996 PMid:22643502 PMCid:PMC3340956
- Al-Jaouni
S, Al-Muhayawi S, Qari M, Nawas MA, Abdel-Razeq H. The safety of
avoiding transfusion preoperatively in patients with sickle cell
hemoglobinopathies.Blood 2002 Nov 16 (Vol. 100, No. 11, pp. 21B-21B).
1900 M STREET. NW SUITE 200, WASHINGTON, DC 20036 USA: AMER SOC
HEMATOLOGY.
- Amar
KO, Rouvillain JL, Loko G. Perioperative transfusion management in
patients with sickle cell anaemia undergoing a total hip arthroplasty.
Is there a role of red-cell exchange transfusion? A retrospective study
in the CHU of Fort-de-France Martinique. Transfusion Clinique et
Biologique. 2013 Mar 1;20(1):30-4. https://doi.org/10.1016/j.tracli.2012.11.001 PMid:23522689
- Marulanda
GA, Minniti CP, Ulrich SD, Seyler TM, Mont MA. Perioperative management
for orthopaedic patients with sickle cell anaemia. Journal of
Orthopaedic Surgery. 2009 Dec;17(3):346-50. https://doi.org/10.1177/230949900901700321 PMid:20065378
- Augier
R, Tennant I, Reid M, Harding H, Crawford-Sykes A, Bortolusso-Ali S,
Isaacs M, Duncan N. Perioperative transfusion of patients with sickle
cell disease undergoing surgery at the University Hospital of the West
Indies (UHWI). The Internet Journal of Anesthesiology. 2008 Volume 21
Number 2
- Bhattacharyya
N, Wayne AS, Kevy SV, Shamberger RC. Perioperative management for
cholecystectomy in sickle cell disease. Journal of Pediatric Surgery.
1993 Jan 1;28(1):72-5. https://doi.org/10.1016/S0022-3468(05)80359-8
- Fu
T, Corrigan NJ, Quinn CT, Rogers ZR, Buchanan GR. Minor elective
surgical procedures using general anesthesia in children with sickle
cell anemia without pre-operative blood transfusion. Pediatric Blood
& Cancer. 2005 Jul 1;45(1):43-7. https://doi.org/10.1002/pbc.20283 PMid:15880471
- Neumayr
L, Koshy M, Haberkern C, Earles AN, Bellevue R, Hassell K, Miller S,
Black D, Vichinsky E. Surgery in patients with hemoglobin SC disease.
American Journal of Hematology. 1998 Feb 1;57(2):101-8. https://doi.org/10.1002/(SICI)1096-8652(199802)57:2<101::AID-AJH2>3.0.CO;2-#
- Firth PG. Anaesthesia for peculiar cells—a century of sickle cell disease. British Journal of Anaesthesia 2005; 95(3): 287-299. https://doi.org/10.1093/bja/aei129 PMid:15863440
- Rubenstein
E. Studies on the relationship of temperature to sickle cell anemia.
The American Journal of Medicine 1961; 30(1): 95-98. https://doi.org/10.1016/0002-9343(61)90066-3
- Edwin
F, Aniteye E, Tamatey M, Frimpong-Boateng K. eComment: Cardiopulmonary
bypass without exchange transfusion in sickle cell disease–An update.
Interactive Cardiovascular and Thoracic Surgery 2010; 10(1): 68-69. https://doi.org/10.1510/icvts.2009.214395A PMid:20019041
- Gross
ML, Schwedler M, Bischoff RJ, Kerstein MD. Impact of anesthetic agents
on patients with sickle cell disease. The American Surgeon 1993; 59(4):
261-264. PMid:8489089
- Yaster
M, Tobin JR, Billett C, Casella JF, Dover G. Epidural analgesia in the
management of severe vaso-occlusive sickle cell crisis. Pediatrics
1994; 93(2): 310-315. PMid:8121746
- Haberkern
CM, Neumayr LD, Orringer EP, et al. Cholecystectomy in sickle cell
anemia patients: perioperative outcome of 364 cases from the National
Preoperative Transfusion Study. Blood 1997, 89(5), 1533-1542.
PMid:9057634
- Tagge
EP, Othersen HB, Jackson SM, et al. Impact of laparoscopic
cholecystectomy on the management of cholelithiasis in children with
sickle cell disease. Journal of Pediatric Surgery 1994; 29(2): 209-213.
https://doi.org/10.1016/0022-3468(94)90320-4
- Al-Mulhim
AS, Al-Mulhim A A. Laparoscopic cholecystectomy in 427 adults with
sickle cell disease: a single-center experience. Surgical Endoscopy
2009; 23(7): 1599-1602. https://doi.org/10.1007/s00464-009-0501-8 PMid:19444510
- Al-Mulhim
AS, Alshehri MH. Laparoscopic cholecystectomy in adult patients with
sickle cell disease. Surgical Laparoscopy Endoscopy & Percutaneous
Techniques 2012; 22(5): 454-458. https://doi.org/10.1097/SLE.0b013e3182619408 PMid:23047392
- Delatte
SJ, Hebra A, Tagge EP, et al. Acute chest syndrome in the postoperative
sickle cell patient. Journal of pediatric surgery 1999; 34(1): 188-192.
https://doi.org/10.1016/S0022-3468(99)90254-3
- Roizen MF. Anesthesia implications of concurrent diseases. In: Miller RD,ed. Anesthesia. 5th ed. Philadelphia: Chuchill Livingstone, 2000; 986.
- Adu-Gyamfi
Y, Sankarankutty M, Marwa S. Use of a tourniquet in patients with
sickle-cell disease. Canadian Journal of Anaesthesia 1993; 40(1),
24-27. https://doi.org/10.1007/BF03009313 PMid:8425239
- Stein
RE, Urbaniak J. Use of the tourniquet during surgery in patients with
sickle cell hemoglobinopathies. Clinical Orthopaedics and Related
Research 1980; 151: 231-233. https://doi.org/10.1097/00003086-198009000-00033
- Al-Ghamdi
AA. Bilateral total knee replacement with tourniquets in a homozygous
sickle cell patient. Anesthesia & Analgesia 2004; 98(2): 543-544. https://doi.org/10.1213/01.ANE.0000099363.42829.0A
- Tobin
JR, Butterworth J. Sickle cell disease: dogma, science, and clinical
care. Anesthesia & Analgesia 2004; 98(2): 283-284. https://doi.org/10.1213/01.ANE.0000099364.07751.3F PMid:14742355
- Fisher
B, Roberts CS. Tourniquet use and sickle cell hemoglobinopathy: how
should we proceed?. Southern medical Journal 2010; 103(11):1156-1160. https://doi.org/10.1097/SMJ.0b013e3181efaf3b PMid:20890260
- Rao
VM, Rao AK, Steiner RM, et al. The effect of ionic and nonionic
contrast media on the sickling phenomenon. Radiology 1982; 144(2):
291-293. https://doi.org/10.1148/radiology.144.2.7089282 PMid:7089282
- Campbell
KL, Hud LM, Adams S, et al. Safety of iodinated intravenous contrast
medium administration in sickle cell disease. The American Journal of
Medicine 2012; 125(1): 100-e11. https://doi.org/10.1016/j.amjmed.2011.06.010 PMid:22195536
- Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood 2012; 120(18): 3647-3656. https://doi.org/10.1182/blood-2012-04-383430 PMid:22923496
- Stein
PD, Beemath A, Meyers FA, Skaf E, Olson RE. Deep venous thrombosis and
pulmonary embolism in hospitalized patients with sickle cell disease.
The American Journal of Medicine 2006; 119(10), 897-e7. https://doi.org/10.1016/j.amjmed.2006.08.015 PMid:17000225
- Ataga
KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious
paradox. The American Journal of Medicine 2003; 115(9): 721-728. https://doi.org/10.1016/j.amjmed.2003.07.011
- Whelihan
MF, Lim MY, Walton BL, et al. Hypercoagulability in Sickle Cell
Disease: The Importance of the Cellular Component of Blood. Blood 2014;
124(21): 4060.
- Galicier
C, Richet H. A prospective study of postoperative fever in a general
surgery department. Infection Control 1985; 6(12): 487-490. https://doi.org/10.1017/S0195941700063608 PMid:3852797
- Pile JC. (2006). Evaluating postoperative fever. Cleveland Clinic Journal of Medicine 2006; 73(1): S62-6. https://doi.org/10.3949/ccjm.73.Suppl_1.S62 PMid:16570551
- Halvorson
DJ, McKie V, McKie K, Ashmore PE, Porubsky ES. Sickle cell disease and
tonsillectomy: preoperative management and postoperative complications.
Archives of Otolaryngology–Head & Neck Surgery 1997; 123(7):
689-692. https://doi.org/10.1001/archotol.1997.01900070033005
- Homi J, Reynolds J, Skinner A, Hanna W, Serjeant G. General anaesthesia in sickle-cell disease. BMJ 1979; 1(6178): 1599-1601. https://doi.org/10.1136/bmj.1.6178.1599 PMid:466140 PMCid:PMC1599173
[TOP]