Kanjaksha Ghosh1, Kanchan Mishra1, Avani Shah1, Parizad Patel1 amd Shrimati Shetty2.
1 Surat
Raktadan Kendra & Research Centre. Udhna Magdalla Road (Nr.
Chosath Joganio Mata Temple), Surat 395002 , Gujrat , India.
2 National Institute of Immunohaematology, 13 the floor KEM hospital MS building, Parel, Mumbai 400012, Maharashtra, India.
Correspondence to: Prof Kanjaksha Ghosh MD, FRCP. Director: Surat Raktadan Kendra & Research Centre. E-mail:
kanjakshaghosh@hotmail.com
Published: March 1, 2019
Received: September 27, 2018
Accepted: January 12, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019018 DOI
10.4084/MJHID.2019.018
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
An
otherwise healthy male child of 9 years presented with paroxysmal fever
and diffuse abdominal pain along with the loss of appetite and nausea
lasting for 3-4days every 4-6 weeks in the last two years. He also has
stretchable skin and hypermobile joints, inherited from his mother who
never suffered any paroxysmal attack of the kind. Work up for acute
intermittent porphyria, lead poisoning, and familial Mediterranean
fever was negative. A novel harmful sequence change in the NLRP12 gene
was detected, and a diagnosis of NLRP12 associated autoinflammatory
syndrome was made. This sequence change within the NLRP12 gene causing
disease has not yet been reported in the literature and is the first
such a case reported from India.
|
Introduction
Mutations in several genes are known to be involved in autoinflammatory syndromes which present in a paroxysmal fashion.[1,2]
These
proteins assemble inside a cell on getting noxious stimulus from the
environment. These assembled proteins produce what is called an
inflammasome. Once inflammasomes are assembled, they activate secretion
of proinflammatory cytokines through various pathways. These cytokines,
e.g., IL-1, TNF-alpha, or IL-6 produce inflammation. A large number of
proteins that go on create inflammasomes have been variously named, and
some of them have multiple domains allowing many proteins to attach and
activate other proteins. Detailed nomenclature of these proteins has
been described elsewhere.[2] Mutations of some of
these proteins lead to the spontaneous assembly of inflammasomes often
under natural environmental conditions or in the cold environment etc.
leading to the autoinflammatory condition.
Clinical presentations
of these conditions vary, i.e., paroxysmal fever, musculoskeletal pain;
skin rashes, abdominal pain, etc. These autoinflammatory syndromes are
caused by mutations in a number of proteins involved in the initiation
and formation of Inflammasomes.[3] NLRP12 is one such protein, and its mutation causes familial cold associated periodic fever syndrome type [2.3,4]
Only a few such cases have been reported in the world literature.[5-7]
We are presenting here another patient with autoinflammatory syndrome
due to a novel heterozygous deleterious sequence change in the NLRP12
gene. The case is being reported with the full consent of the parents
of the patient as the patient is minor.
The Case
A 9-year-old boy (45 kg,1.49 mts), full-term normal delivery, 2nd
child in the family, born of the nonconsanguineous marriage, having
good intelligence (std 5, rank holder in the class), completed all the
immunization without any complication and was apparently healthy. He
presented with recurrent episodes of abdominal pain, moderate grade
fever (37.5-38°C), complete loss of appetite with nausea but no
vomiting for last two years along with a feeling of soreness all over
the body. This paroxysmal presentation was coming on once every 4-6
weeks without any apparent relation to food, weather or other
environmental or extraneous factors. The paroxysm lasted for 3-4 days,
without any skin rash, aphthous ulcers in the mouth, or a sore throat.
There were no cold sores. He did not have any skin rash or skin ulcers.
There were no sore throat or lymphadenopathy. He was clinically
examined during 3 of his attacks when he was found to be withdrawn,
apparently suffering continuous pain by his expression. His clinical
examination showed a well-built child with hyper stretchable skin, fish
mouth scars, and hypermobile joints. There was no iridodonesis, and his
vision was normal (6/6 both eyes), there was no other suggestion of
Marfan’s syndrome on various measurements of the body. Mild diffuse
tenderness was found all over the abdomen with normal bowel sounds.
There was no organomegaly, and the pain was poorly localized. There
were no cardiac murmur, respiratory abnormality, focal neurological
abnormality, and the rest of the clinical examination was essentially
normal. He recovered automatically after 3-4 days of the ordeal.
He had history neither of any serious illness nor of food or drug allergy.
Elder
brother (14 years of age) and parents had no such illness, but the
mother has lax joints and skin. There were no pets in the house and no
family history of a similar disease in the extended family.
His
complete blood count showed neutrophilic leukocytosis (TLC 14-15000/ul
with 76 percent polymorphs and no eosinophils during the acute stage)
with normal hemoglobin and platelet counts. There were no abnormalities
in the morphology of any of the cells seen. His biochemical
investigations which involved the liver, renal function tests,
electrolytes also showed no abnormality. Blood and urine culture and
routine urine examination were unremarkable. Urine for porphobilinogen
showed a mild increase (Figure 1)
however the quantitation of PBG (porphobilinogen) on 24-hour urine
during acute stage was normal so also the blood lead levels
(<10ugm/L). Serum amylase, lipase, and lipid profile were normal.
CRP levels were raised during the acute stage (170mg/L) and was quickly
normalized (3mg/L) within 72 hours of resolution of abdominal pain.
Upper GI endoscopy and fundoscopy (eye) showed no abnormality. Serum
immunoglobulins including IgD levels were normal. Ultrasound
examination of abdomen and pelvis, CT scan with contrast and MRI scan
of chest and abdomen was essentially normal.
 |
Figure
1. Urine showing mild increase in porphobilinogen( Hoesch test) and
partitioning with chloroform in urine of the patient (ruby red color). |
Because
of the paroxysmal nature of the attack, a provisional diagnosis of
acute intermittent porphyria or one of the autoinflammatory syndromes
was made.
Targeted sequencing of exome involving porphyrin
metabolism, autoinflammatory conditions, and immunologically important
280 genes was executed following transposase digestion of isolated DNA
from the peripheral blood mononuclear cells of the patient on Illumina
2500 Hi seq platform. Except for nine genes (CD55, CFHR1, CORO1A,
ITCH1, MAGT1, TNFRS11A, TBX1, FCGR1A, NCF1, 85-98 % coverage), all
genes were covered to the tune of 100% of the exomes including
intron-exon boundaries.
Except for common polymorphisms, none of
the genes showed any harmful sequence changes or known mutations.
However, there was a heterozygous mutation of NLRP12 gene in exon 3 (c
779C>T, p Thr 260> Meth) in the evolutionarily conserved
nucleotide sequence on NACHT1 domain of the molecule. This sequence
change has not been described previously and was found to be harmful by
using Polyphen 2 Sift, Mutation tester-2 software.
This sequence
change was confirmed using Sanger sequencing but was not found in any
of the parents or his elder brothers DNA sequences. Hence it presented
a de novo change. So a final diagnosis of NLRP12 associated
autoinflammatory syndrome was made. The patient did not respond to
colchicine, and by trial and error with various combinations of
anti-inflammatory medicines, Naproxen gave a partial response with a
combination of a short course of corticosteroids (Prednisolone, 15
mg/day, administered when paroxysms started once in the morning after
breakfast and was continued for seven days.
Discussion
NLRP12
gene product is a member of the CATERPILLER family of cytoplasmic
proteins. This protein contains an N-terminal pyrin domain, a NACHT
domain, a NACHT-associated domain, and a C-terminus leucine-rich repeat
region(LRR) which functions as an attenuating factor of inflammation by
suppressing inflammatory responses in activated monocytes. Mutations in
this gene cause familial cold autoinflammatory syndrome type 2.
Alternative
splicing of the mRNA of this gene results in multiple transcript
variants in different tissues, and which may be responsible for the
presentation in different organs with related symptoms.
In the
absence or reduced function in different domains of the protein
inflammatory cytokine IL1 beta is produced in excess via an increase in
NFkbeta activity and is responsible for many of the inflammatory signs
and symptoms of the disease.[1,8]
There
are only a few cases (forty-four till date) of paroxysmal
autoinflammatory syndrome reported with NLRP12 mutation. This condition
with autosomal dominant inheritance normally presents with paroxysmal
cold-induced periodic fever syndrome with cutaneous, musculoskeletal,
lymph node related symptoms.[3,5,6,8] A patient with common variable immunodeficiency associated with this mutation has also been reported.[7]
Our
patient, however, had several interesting features ie the pathology
presented not in early childhood but later. He had no cutaneous
symptoms peculiar to NLRP12 mutation, but he had joint laxity,
stretchable skin and fishmouth scars like pseudoxanthoma elasticum
which may be unrelated to this mutation. He clearly inherited from his
mother stretchable skin and hypermobile joints as she also had similar
features, but she did not have any symptoms of NLRP12 mutation.
The
reason why the patient did not get any cutaneous manifestation, known
to occur with the disease may be related to the tropical high ambient
temperature in the city of Surat where the boy lived.
Cutis laxa (pseudoxanthoma elasticum) can be caused by mutations in several genes, including ATP6V0A2, ATP7A, EFEMP2, ELN, and FBLN5.
Most of these genes are involved in the formation and function of
elastic fibers, which are slender bundles of proteins that provide
strength and flexibility to connective tissue
throughout the body. There were no mutation or sequence changes in any
of these genes in our patient, neither was there any mutation detected
in the porphyrin metabolism genes to account for this paroxysmal pain
and fever.
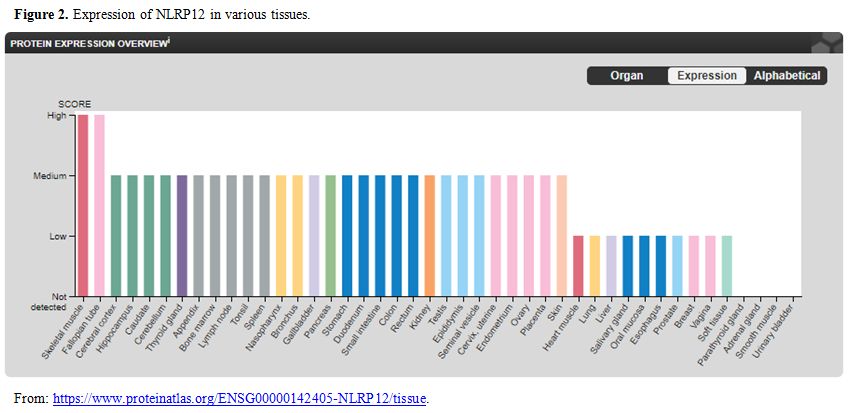
The present case is a novel de-novo NLRP12 mutation as
it was not found in any of the parents or his elder brother. NLRP12
gene is expressed in many tissues (Figure 2).
Hence depending on local tissue pathology recurrent inflammation can
take place in any tissue and explain the protean nature of
manifestation in this disease. Till date in the world literature, only
44 cases of NLRP12 mutation associated autoinflammatory syndrome has
been reported. A good number (fifteen) of them has been reported
amongst a large cohort of patient with suspected immunodeficiency,[4]
when the next-generation sequencing was applied to look for the cause
of autoinflammatory syndrome or to rule out a familial Mediterranean
fever, a well-known cause of the inflammatory syndrome. The authors and
the milder presentation have well-described heterogeneity of
presentation with this mutation compared to NLRP3 mutation has been
emphasized.[4] In 44-50% of these patients a F402L
mutation has been commonly described. Nonsense changes in NLRP12 gene
pose no problem in describing it as the genuine pathogenic cause of the
disease but a missense mutation needs more attention if it is a single
case reported from the world and similar missense mutation from other
patients are lacking as has happened in the present case.
In
such a situation the change needs to be noted and whether the change is
deleterious needs to be confirmed through standard software analysis.
The mutation described in the present paper was found to be deleterious
using more than one software as already described, and this change is
in NACHT domain of the protein which has several important functions
like NTP use activity, nucleotide binding activity, etc. to name a few.
Several mutations (e.g., D294E, R211H, Y246C, R352C, H304Y) in NACHT
domain (aa 211 to aa391) has been reported by other authors either as
single or two cases and was pathogenic when analyzed on several
softwares as described in this paper.[5,7]
Management of this condition has not been standardized, but a good
proportion of patients responded to corticosteroids, NSAIDs and
anti-allergic drugs given alone or in various combinations. Some of the
patients need therapy directed to TNF alpha or Interleukin 1 beta in
the form of adalimumab, Canakinumab,
infliximab. These antibodies neutralize inflammatory mediators like
IL-1, TNF alpha or IL-6 which is increased in this condition and is
known to be the final pathogenic mediator in this condition. A few
patients have developed amyloidosis or Crohn’s disease on follow up.[5,9,10]
A recently published review has provided very good information on the
management clinical presentation and diagnosis of the systemic
autoinflammatory disorder in children.[11]
From
India To date, no case of NLRP12 related autoinflammatory disorder has
been reported, making this the first such a case from this country.
References
- Manthiram K, Zhou Q, Aksentijevich I, Kastner DL.
The monogenic autoinflammatory diseases define new pathways in human
innate immunity and inflammation. Nature Immunology 2017, 18: 832-42. https://doi.org/10.1038/ni.3777 PMid:28722725
- Rigante
D. A systemic approach to autoinflammatory syndromes: A spelling
booklet for the beginner. Expert. Rev. Clin. Immunol. 2017,13:571-97. https://doi.org/10.1080/1744666X.2017.1280396 PMid:28064547
- Broderick
L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and
autoinflammatory syndromes.Annu Rev Pathol. 2015;10:395-424. https://doi.org/10.1146/annurev-pathol-012414-040431 PMid:25423351
- Jeru
I, Le Borgne G, Cochet E, Hayrapetyan H, Duquesnoy P, et al.
Identification and functional consequences of a recurrent NLRP12
missense mutation in periodic fever syndromes. Arthritis Rheum.
2011;63:1459–64. https://doi.org/10.1002/art.30241 PMid:21538323
- Kostik
MM, Suspitsin EN, Guseva MN, Levina As, Kazantseva Ay, Sokolenko AP,
Imyanitov En. Multigene sequencing reveals heterogeneity of NLRP12
related autoinflammatory disorders. Rheum Internat. 2018. https://doi.org/10.1007/s00296-018-4002-8. Published on line March 2018.
- Vitale
A, Rigante D, Maggio MC, Emmi G, Romano M, Silvestri E, Lucherini OM,
Emmi L, Gerloni V, Cantarini L. Rare NLRP12 variants associated with
the NLRP12-autoinflammatory disorder phenotype: an Italian case series.
Clin Exp Rheumatol. 2013;31(3 Suppl 77):155-6. PMid:24064030
- Shen
M, Tang L, Shi X, Zeng X, Yao Q. NLRP12 autoinflammatory diseases: a
Chinese case series and literature review. Clin Rheumatol. 2016,
https://doi.org/10.1007/s10067-016-3410-y
- Lupfer C, Kanneganti T- D. Unsolved mysteries in NLR biology. Front. Immunol. 2013; 4 (art 285). 1-10.
- Borte
S, Celiksoy MH, Menzel V, Ozkaya O , Ozen FZ, Hammarström L, Yildiran
A. Novel NLRP12 mutations associated with intestinal amyloidosis in a
patient diagnosed with common variable immunodeficiency. Clin Immunol.
2014 ;154:105-11. https://doi.org/10.1016/j.clim.2014.07.003 PMid:25064839
- Terhar
N, Lachmann H, Ozen S, Woo P, Uziel Y, Modesto C et al. Treatment of
autoinflammatory diseases: results from Eurofever Registry and review.
Ann Rheum Dis. 2013;72:678-85. https://doi.org/10.1136/annrheumdis-2011-201268 PMid:22753383
- Rigante.
D. The broad-ranging panorama of systemic autoinflammatory disorders
with specific focus on acute painful symptoms and hematological
manifestations in children. Mediterr. J. Hematol. Infect Dis. 2018; 10
(1) :e2018067. DOI: https://doi.org/10.4084/mjhid.2018.067
[TOP]
{kind=link}