Tite Minga Mikobi1,2, Prosper Tshilobo Lukusa1,3, Jean-Marie Mbuyi Muamba4 and Tozin Rhama5.
1 Center for Human Genetics, Faculty of Medicine, University of Kinshasa, Democratic Republic of Congo, DRC.
2 Division of Gynecology Obstetrics, Center for Sickle Cell Anemia, Kinshasa, DRC.
3 Division of Pediatrics, Hospital University, Faculty of Medicine, University of Kinshasa, DRC.
4
Division of Internal Medicine, Service of Immuno hemato Rheumatology,
Hospital University, Faculty of Medicine, University of Kinshasa, DRC.
5 Division of Gynecology Obstetrics, Hospital University, Faculty of Medicine, University of Kinshasa, DRC.
Correspondence to: Tite Minga Mikobi. Center for Human Genetics,
Faculty of Medicine, University of Kinshasa, Democratic Republic of
Congo, DRC. E-mail:
tite.mikobi@unikin.ac.cd
Published: July 1, 2019
Received: December 18, 2018
Accepted: May 22, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019039 DOI
10.4084/MJHID.2019.039
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Objective:
to determine the beneficial role of Fetal Hemoglobin (FHb) and
alpha-thal on fetal and maternal morbidity during pregnancy in sickle
cell patients.
Study site: the study was conducted at the sickle cell center of Kinshasa between 2008 and 2018
Setting and study population: this is a documentary and analytical study that included 980 deliveries of homozygous sickle cell patients.
Methods:
the diagnosis of SCD and the quantification of FHb were performed with
the capillary electrophoresis technique. The molecular test confirmed
the diagnosis of SCD. The diagnosis of alpha-thal was made with the
multiplex ligation-dependent probe amplification (MLPA) technique.
Sickle cell pregnancies were followed according to the protocol of care
in force in the University of Kinshasa Hospital service. The variables
of interest were: hematological variables, sickle cell crises during
pregnancy, maternal and fetal complications.
Statistics:
statistical analyses were performed with SPSS 20.0 software. Means and
standard deviations were compared with the Student's t and ANOVA tests.
The value of p <0.05 was considered the significance level.
Results:
the Hb-SS / alpha-thal and HbSS / HPFH genotypes were observed in 101
and 121 women, respectively. Otherwise, 758 women had HbSS genotype.
The morbidity related to sickle cell complications in the mother and
fetus were less frequent in the Hb-SS / alpha-thal and HbSS / HPFH
groups than in HB-SS group. The differences were statistically
significant.
Conclusion: this study showed a significant protective effect of alpha-thal and HPFH during pregnancy in sickle-cell pregnant women.
|
Introduction
Sickle
cell disease (SCD) is a constitutional hemoglobinopathy with autosomal
recessive inheritance. The characteristic of this mutation is a
transversion of a purine base [A] by a pyrimidine base in the
beta-globin gene. The consequence of this mutation is the substitution
of glutamic acid by valine at position 6 in the beta globin chain.[1,2]
The substitution of a hydrophilic amino acid (glutamic acid) by a
hydrophobic amino acid (valine) results in the production of abnormal
hemoglobin called HbS. Indeed, in concentrated solution and under the
influence of a decrease in oxygen partial pressure, HbS undergoes a
supramolecular polymerization process.[3] Hemoglobin S is today the most widespread structural abnormality of hemoglobins in the world.[4,5]
Equatorial Africa is the area of maximum incidence. Clinically SCD is
characterized by recurrent vaso-occlusive ischemic events, chronic
hemolysis, and high susceptibility to infections. The best management
begins with early detection, preventive care against encapsulated
bacterial infections and especially the administration of hydroxyurea.
The association of SCD and pregnancy is characterized by high maternal
and fetal morbidity.[6,7] Indeed, it is well
established that there is a reciprocal influence between pregnancy and
SCD. During pregnancy, there is a worsening of maternal anemia, an
increase in the frequency of VOC, and a high risk of infection.[8]
The fetus is at high risk of spontaneous abortions, prematurity,
intrauterine growth retardation, hypotrophy, and in utero death.[9-11] Genetically, two factors showed their modulatory effect on the sickle cell phenotype. These are fetal hemoglobin (FHb: α2γ2)
and alpha-thalassemia. A high level of FHb slows down the
polymerization, which results in a reduction of the number of VOC and
hemolysis. The mechanism of this protection is explained by the
formation of hybrid polymers (α2βsγ)
that stop the growth of the phenomenon of polymerization. This property
has since been used in the treatment of SCD following empirical
evidence that hydroxyurea stimulated the production of FHb.[12,13]
The protective effect of alpha-thalassemia is related to the reduction
of Hb concentration in the erythrocytes, which results in the
microcytic anemia.[14] Indeed, Mikobi et al. showed
that homozygous alpha-thalassemia and an FHb level > 15% had a
protective effect on SCD in Congolese patients.[15]
The objective of the present study was to determine the beneficial role
of genetic modulating factors of sickle cell disease in fetal and
maternal morbidity and mortality during pregnancy in sickle cell
patients.
Patients and Methods
Subjects.
In this is a transversal study, 980 records of SCD pregnant women,
who gave birth between 2008 and 2018, were analyzed. The review was
conducted at the Sickel Cell Center in Kinshasa, Democratic Republic of
Congo (DRC). The patients were divided into three genotypic subgroups.
The first subgroup consisted of SCD pregnant with associated homozygous
alpha-thalassemia (Hb-SS/alpha-thal), the second group consisted of SCD
with hereditary persistence of fetal hemoglobin (HbSS / HPFH), and the
third group consisted of gestants without association with one of the
two pathologies mentioned above (HbSS).
Operational definitions.
In this study, the group of gestational Hb-SS /alpha-thal included only
patients who had the deletion alpha-thalassemia homozygous (-α, -α or
--, αα) and the group HbSS / HPFH the patients with an FHb level >
15%. However, all sickle cell patients with heterozygous
alpha-thalassemia (-α, αα or αα, -α) or with FHb < 15% were excluded
from this study.
Laboratory tests.
The diagnosis of SCD was made by the technique of capillary
electrophoresis; the device used was the Mini cap flex piercing (Sebia,
France). This technique also made it possible to quantify the fraction
of FHb and to make the diagnosis of HPFH. The diagnosis of HPFH was
retained for an FHb > 15% after three dosages within three months.
The diagnosis of SCD was confirmed by a molecular test based on the
restriction fragment-length polymerization technique (RFLP). The
diagnosis of alpha-thalassemia has been made by the Multiplex
Ligation-dependent Probe Amplification (MLPA) technique. The
procedures, as well as the reagents used for both techniques (RFLP and
MLPA), have been carefully described by Mikobi et al..[15,16]
Protocol for the management of sickle cell disease.
Transfusion and pain management.
Our protocol advocates only therapeutic transfusions during pregnancy.
Prophylactic transfusions which systematically transfuse sickle cell
disease from the 24th weeks of
pregnancy are not practiced in our department for two main reasons:
economic and lack of consensus in the real benefit of systemic
prophylactic transfusion. However, we will resort to occasional
transfusion exchanges during the pregnancy when there is an indication.
Vaso-occlusive pain at levels 1 and 2 was managed with analgesics
according to the recommendations of the WHO. Complicated VOC with Stage
3 pain was managed with a multidisciplinary team consisting of
anesthesia intensive care and hematologist.
Maternal pregnancy supplements and fetal monitoring.
All pregnant women were given a systematic supplementation of iron and
folic acid, and anti-oxidants (omega 3 fatty acids and magnesium
pidolate) from the 12th week of amenorrhea. Prophylactic anti-malarial treatment was given between the 24th and 32nd
weeks, and anti-helminthic deworming was given between 28 and 32 weeks.
No patients received hydroxyurea during pregnancy. Fetal surveillance
was primarily based on ultrasound scans.
Childbirth. Delivery was systematically scheduled for the 37th week of amenorrhea after the completion of the biophysical manning score.
Variables of interest for the study.
In this study, the following general maternal variables were assessed:
gestational age, menarche age, parity, weight gain. The pregnancy
weight gain (ΔP) was calculated from the following formula: ΔP = Pf -
Pi (Pf weight of the pregnant woman at the time of delivery, Pi weight
before pregnancy). The evolution of the Hb rate allowed to appreciate
the validity of a punctual transfusion. Maternal morbidity was assessed
by the analysis of sickle cell complications: VOC and hemolysis as a
function of gestational age, including pregnancy complications:
preeclampsia, acute chest syndrome (ACS), parasitic infections
(malaria), bacterial infections (urinary tract infections, pneumonia,
sepsis) and postpartum, endometritis. Fetal morbidity was assessed by
analysis of abortion rates, prematurity, low birth weight, and in utero
death.
Statistical analyses.
Statistical analyses were performed using SPSS version 20.0.T. (2016).
We had determined the distribution of the study population, which was
normal. We have determined also means and standard deviations. Mean of
two groups were compared by the Student's t-test and those of three
groups by the ANOVA test. The value of p <0.05 was considered the
only one of significance.
Results
Our
study showed that 101 (10.30%) of our sickle cell deliveries had a
homozygous alpha-thal deletion, while 121 (12.34%) of the women had an
HPFH. Besides, 758 or 77.34% of the deliveries had none of the two
associated genetic factors.
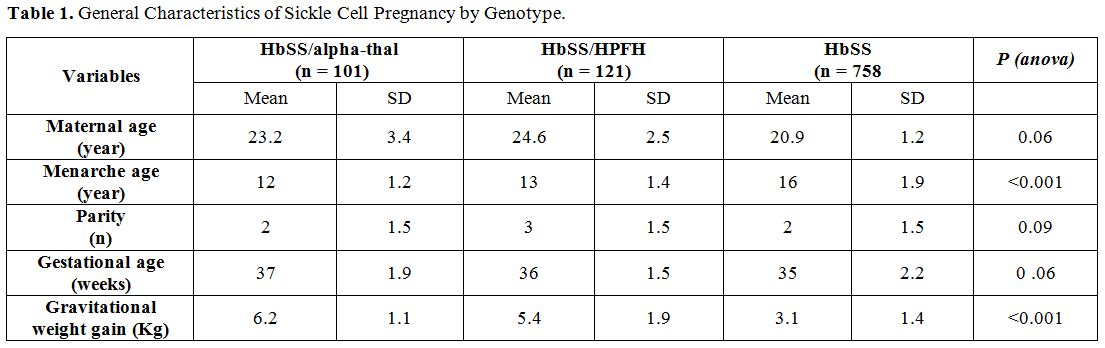
Table 1
gives the general characteristics of sickle-cell pregnancies according
to their genotype. The analysis in the table shows that the Hb-SS /
alpha-thal and HbSS / HPFH women had their menarche before those of the
HbSS genotype. In addition, these gestants (HbSS/alpha-thal and
HbSS/HPFH) had a better weight gain. Statistical differences between
the first two groups and the third group were highly significant (p
<0.001).
 |
Table
1. General Characteristics of Sickle Cell Pregnancy by Genotype. |
Table 2
shows the biological variables during pregnancy. From the analysis in
this table, the gestants of the HbSS genotype had higher levels of WBC,
platelets, and reticulocytes than those with an alpha-thal or HPFH
combination. The differences were highly significant (p<0.001). In
contrast, serum iron and Hb were similar in all three groups.
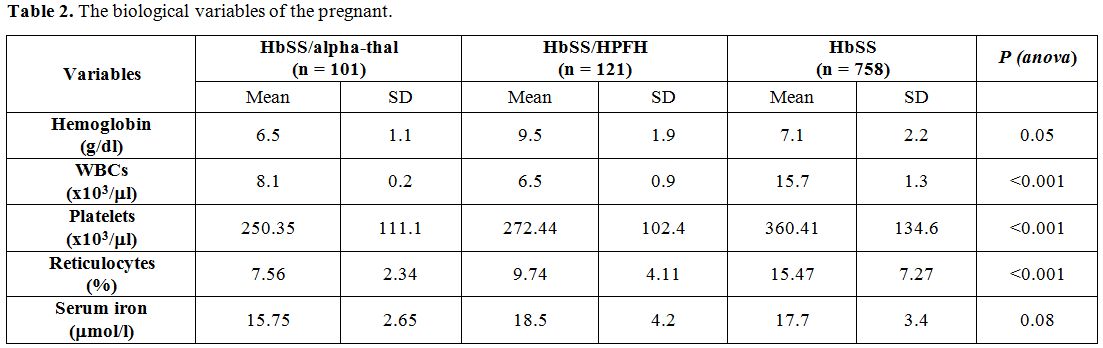 |
Table 2. The biological variables of the pregnant. |
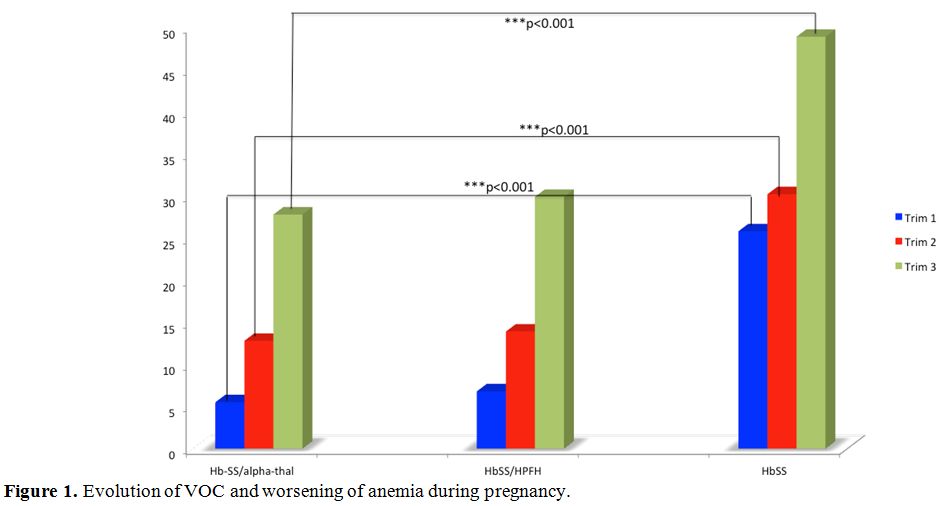
Figure 1
shows the distribution of sickle cell crises during pregnancy. The
chart analysis shows that VOC and hemolysis increase steadily with
gestational age. They reach their maximum in the third trimester.
However, the HbSS genotype is more affected than the other two groups.
Statistical differences with the Hb-SS/alpha-thal group are highly
significant (p<0.001).
 |
Figure
1. Evolution of VOC and worsening of anemia during pregnancy. |
Table 3
presents the frequencies of maternal and fetal complications. The
analysis in this table shows that the frequencies of ACS and
pre-eclampsia were similar in all three groups. In contrast,
spontaneous abortions, prematurity, low birth weight, in utero death
and maternal death have been more observed in the HbSS genotype.
Statistical differences with the other two genotypes were highly
significant (p <0.001).
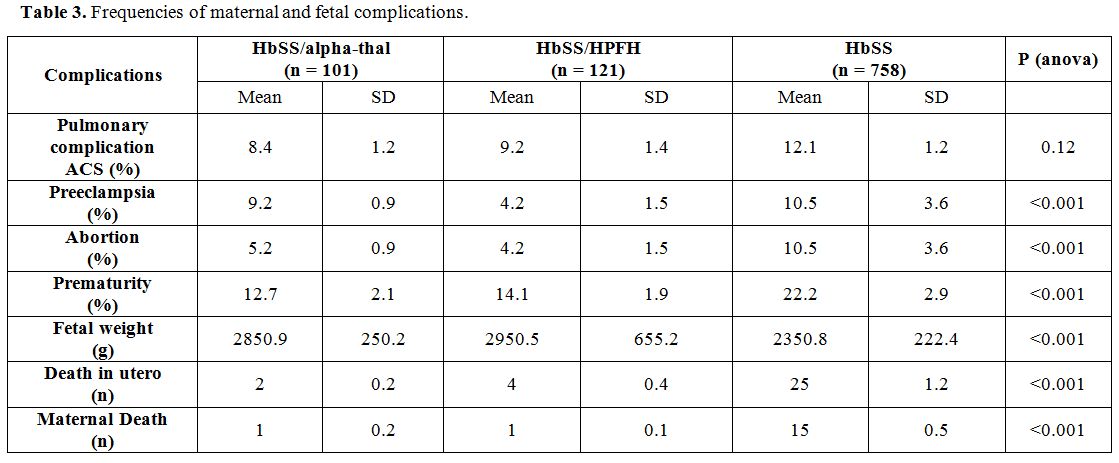 |
Table 3. Frequencies of maternal and fetal complications. |
Figure 2
shows the distribution of parasitic and bacterial infections during
pregnancy. It appears that the frequencies of malaria were similar in
the three groups. In contrast, bacterial infections (urinary tract
infections, pneumonia, sepsis, and endometritis) were more common in
the HbSS genotype. The statistical differences were highly significant
(p <0.001).
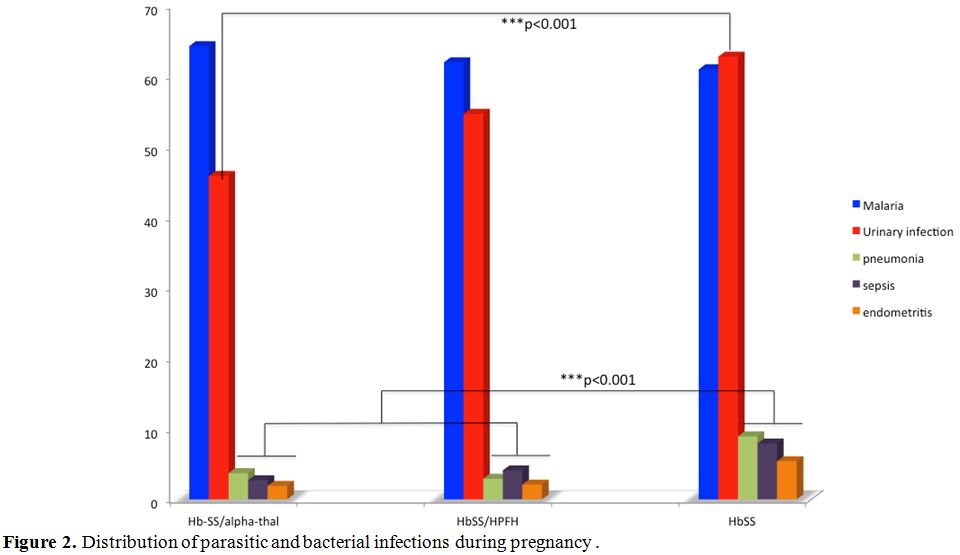 |
Figure 2. Distribution of parasitic and bacterial infections during pregnancy. |
Delivery route.
In our series, 49% of Caesarean sections were performed in patients
with the HbSS genotype. Whereas the rates of the cesarean section of
pregnant women with the HbSS / alpha and HbSS / HbF genotypes were 24%
and 27% respectively.
Discussion
FHb and alpha-thal are recognized as modulatory factors for the clinical expression of SCD.[17] However, their beneficial effect during pregnancy is not well known in subtropical Africa.
In
this study, sickle cell patients with HbSS genotypes had their menarche
late at 16 years of age. Puberty delay is usually observed in SCD and
is proportional to the severity of the disease.[18] The delivery was programmed at the 37th week; this attitude is the one reported by many authors.[19,20]
Our study showed a significant difference in weight gain in favor of
the HbSS / PHHF and HbSS / alpha-thal genotypes. The observed
difference could be associated with the protective effects of HbF[21] and alpha-thal.[22]
From the hematological variables, the HbSS genotype has a high number
of WBCs, reticulocytes, and platelets. These high biological variables
can explain the high morbidity of these patients during pregnancy.
VOC episodes were the leading cause of morbidity during pregnancy in SCD, as also reported by other authors.[23]
These VOC increase with gestational age and are more frequent and
severe in the perinatal period. In our series, VOC seizures were more
common in the HbSS group. During pregnancy, several authors report a
high frequency of complications such as acute thoracic syndrome and
pre-eclampsia.[23,24] Their frequencies (9 to 16%) vary from one series to another.[20,23,24]
In our series, the frequencies of these two complications are similar
to those reported by other authors on the one hand and similar between
the three genotypes on the other hand. During pregnancy, spontaneous
abortions, prematurity, hypotrophy, and in utero fetal death are the
main complications found in SCD.[20,25]
The frequencies are different from one series to another; they are also
proportional to the severity of the disease during pregnancy. In our
series, these complications are more common in the HbSS group.
Infection
is ranked as the second leading cause of morbidity during pregnancy in
SCD. In our series, malaria was the first parasitic infection
encountered because of the geographical situation of DRC. Indeed, DRC
is located in the area with the highest prevalence of malaria. This
infection alone constitutes the first cause of maternal and fetal
morbidity.[26] In tropical Africa, malaria is ranked
as the leading cause of fetal hypotrophy. The high morbidity of malaria
during pregnancy is associated with maternal anemia that Plasmodium
falciparum can cause.[27] In our series, the
frequency of malaria was similar in all three genotypes. Maternal
morbidity has also been influenced by bacterial infections such as
urinary tract infection, pneumonia, sepsis, and endometritis. Their
frequencies are similar to those reported by other authors.[20]
However, in our series, these infections were more common in the HbSS
group. The high frequency of cesarean section in SCD is related to
peripartum complications. In our series, 49% of pregnant women had
delivered by cesarean section. This frequency is similar to those
reported by other authors.[20] However, the group
with the HbSS genotype was more exposed to cesarean section than the
other two. The protective effect of HbF and alpha-thal observed in our
series, is similar to that reported by other authors in the
associations S/β(0) thalassemia, S/β(+) thalassemia,[21] or other major sickle cell syndromes like SC, SD.
Conclusions
Homozygous
alpha-thal and PHHF have shown their protective effect on sickle cell
disease in general. This study shows that these genetic factors
modulating sickle cell phenotype can significantly reduce maternal and
fetal morbidity during pregnancy.
References
- Ingram VM. A specific chemical difference between
the globins of normal human and sickle cell anemia hemoglobin. Nature,
1956; 178: 792-4. https://doi.org/10.1038/178792a0 PMid:13369537
- Ingram
VM. Abnormal hemoglobin. The chemical difference between normal and
sickle cell hemoglobins. Biochim - Biophys Acta, 1959; 36: 402-411. https://doi.org/10.1016/0006-3002(59)90183-0
- Perutz MF, Mitchison JN. State of hemoglobin in sickle cell anemia. Nature, 1950; 166: 677-679. https://doi.org/10.1038/166677a0
- Serjeant GR - Sickle cell disease. Lancet, 1997; 350: 725-730. https://doi.org/10.1016/S0140-6736(97)07330-3
- Serjeant GR. The Natural History of Sickle Cell Disease. Cold Spring Harb Perspect Med, 2013; 3:a011783 https://doi.org/10.1101/cshperspect.a011783 PMid:23813607 PMCid:PMC3784812
- Hendrickse
JPdeV, Harrison KA, Watson-Williams EJ, Luzzatto L, Ajabor LN.
Pregnancy in homozygous sickle-cell anemia. J Obstet Gynecol Br Com-
monw 1972;79:396-409. https://doi.org/10.1111/j.1471-0528.1972.tb14177.x
- Baum
KF, Dunn DT, Maude GH, Serjeant GR. The painful crisis of homozygous
sickle cell disease. A study of risk factors. Arch Intern Med
1987;147:1231-4. https://doi.org/10.1001/archinte.1987.00370070045007 PMid:3606281
- Koshy M, Burd L. Management of pregnancy in sickle cell syndrome. Hematol Oncol North Am 1991;5(3):585-96. https://doi.org/10.1016/S0889-8588(18)30433-7
- Powars DR, Sandhu M, Niland-Weiss J et al. Pregnancy in SSD. Obstet Gynecol 1986; 67:217-28. https://doi.org/10.1097/00006250-198602000-00012 PMid:3945432
- Sun
PM, Wilburn W, Raynor D et al. SSD in pregnancy: twenty years of
experience at Grady Memorial Hospital, Atlanta, Georgia. Am J Obstet
Gynecol 2001;184:112-30. https://doi.org/10.1067/mob.2001.115477 PMid:11349177
- Serjeant GR, Loy LL, Crowther M et al. Outcome of pregnancy in homozygous SSD. Obstet Gynecol 2004;103(6):1278-85. https://doi.org/10.1097/01.AOG.0000127433.23611.54 PMid:15172865
- Nagel
RL, Bookchim RM, Johnson J et al. Structural bases of the inhibitory
effects of hemoglobin F and hemoglobin A2 on the polymerization of
hemoglobin S. Proc Natl Acad Sci USA, 1982;76: 670 - 2. https://doi.org/10.1073/pnas.76.2.670 PMid:284392 PMCid:PMC383012
- Cannas
G, Poutrel S, Thomas X. Hydroxycarbamine: from an Old Drug Used in
Malignant Hemopathies to a Current Standard in Sickle Cell Disease.
Mediterr J Hematol Infect Dis. 2017 Feb 15;9(1):e2017015. doi:
10.4084/MJHID.2017.015. eCollection 2017. Review. https://doi.org/10.4084/mjhid.2017.015 PMid:28293403 PMCid:PMC5333733
- Higgs
DR, Aldridge BE, Lamb J et al. The interaction of alpha-thalassemia and
homozygous sickle cell disease. N Engl J Med., 1982; 306: 1441 - 6. https://doi.org/10.1056/NEJM198206173062402 PMid:6176865
- Mikobi
TM, Lukusa PT, Aloni MN, et al. Association between sickle cell anemia
and alpha thalassemia reveals a high prevalence of the α3.7
triplication in congolese patients than in worldwide series. J Clin Lab
Anal. 2017;00:e22186. https://doi.org/10.1002/jcla.22186 PMid:28276593
- Mikobi
TM, Lukusa Tshilobo P, Aloni MN, Akilimali PZ, Mvumbi-Lelo G, and
Mbuyi-Muamba JM. Clinical phenotypes and the biological parameters of
Congolese patients suffering from sickle cell anemia: A first report
from Central Africa. J Clin Lab Anal. 2017;00:e22140. https://doi.org/10.1002/jcla.22140 PMid:28116772
- Jit
BP, Mohanty PK, Purohit P, Patel S, Meher S, Mohanty JR, Sinha S,
Behera RK, Das P. Association of fetal hemoglobin level with frequency
of acute pain episodes in sickle cell disease (HbS-only phenotype)
patients. Blood cells Mol Dis. 2019 Mar ;75 :30-34. Epub 2018 Dec 20. https://doi.org/10.1016/j.bcmd.2018.12.003 PMid:30597429
- Yacobovich
J, Tamary H : Thalassemia major and sickle cell disease in adolescents
and young adults. Acta Haematol. 2014;132(3-4) :340-7. Epub 2014 Sep
10. https://doi.org/10.1159/000360235 PMid:25228560
- Chang
JN, Magann EF, Novotny SA, Cooley CE, Gauss CH, Parrish MR, Morrison
JC. Maternal/Perinatal Outcome in Women with Sickle Cell Disease: A
Comparison of Two Time Periods. South Med J. 2018 Dec ;111(12)
:742-745. https://doi.org/10.14423/SMJ.0000000000000900 PMid:30512127
- Silva-Pinto
AC, de Oliveira Domingues Ladeira S, Brunetta DM, De Santis GC, de
Lucena Angulo I, Covas DT. Sickle cell disease and pregnancy: analysis
of 34 patients followed at the Regional Blood Center of Ribeirão Preto,
Brazil. Rev Bras Hematol Hemoter. 2014 Sep-Oct;36(5):329-33. Epub 2014
Jul 16. https://doi.org/10.1016/j.bjhh.2014.07.002 PMid:25305164 PMCid:PMC4318372
- Sokolova
A, Mararenko A, Rozin A, Podrumar A, Gotlieb V. Hereditary persistence
of hemoglobin F is protective against red cell sickling. A case report
and brief review. Hematol Oncol Stem Cell Ther. 2017 Oct 16. pii:
S1658-3876(17)30115-2. https://doi.org/10.1016/j.hemonc.2017.09.003 PMid:29079125
- Resende
Cardoso PS, Lopes Pessoa de Aguiar RA, Viana MB. Clinical complications
in pregnant women with sickle cell disease: prospective study of
factors predicting maternal death or near miss. Rev Bras Hematol
Hemoter. 2014 Jul-Aug; 36(4):256-63. Epub 2014 May 29. https://doi.org/10.1016/j.bjhh.2014.05.007 PMid:25031164
- Cardosa
D, Ridout A, Nanda S, Howard J, Robinson SE, Oteng-Ntim E. Maternal
sickle cell disease and twin pregnancy: a case series and review of the
literature. Hematology. 2019 Dec;24(1):148-158. Epub 2018 Oct 21. https://doi.org/10.1080/10245332.2018.1535534 PMid:30345909
- Chambers
J, Smith N, Sehring M, Chittivelu S. Acute Chest Syndrome Progressing
to ARDS in a Patient of 25-Week Gestation. Case Rep Crit Care. 2018 Jan
30;2018:4243569. eCollection 2018. https://doi.org/10.1155/2018/4243569 PMid:29666710 PMCid:PMC5831955
- Burgos
Luna JM, Páez Rúa DM, Ruiz Ordoñez I, Fernández PA, Escobar Vidarte MF.
Description of criteria for near miss in high-complexity obstetric
population with sickle cell anemia: an observational study. J Matern
Fetal Neonatal Med. 2018 Sep 19:1-6. https://doi.org/10.1080/14767058.2018.1510912 PMid:30231783
- McGann
PT, Williams AM, Ellis G, McElhinney KE, Romano L, Woodall J, Howard
TA, Tegha G, Krysiak R, Lark RM, Ander EL, Mapango C, Ataga KI, Gopal
S, Key NS, Ware RE, Suchdev PS. Prevalence of inherited blood disorders
and associations with malaria and anemia in Malawian children. Blood
Adv. 2018 Nov 13;2(21):3035-3044. https://doi.org/10.1182/bloodadvances.2018023069 PMid:30425067 PMCid:PMC6234379
- Maier AG, Matuschewski K, Zhang M, Rug M. Plasmodium falciparum. Trends Parasitol. 2018 Dec 27. pii: S1471-4922(18)30248-4.
[TOP]