Valder R. Arruda1,2,3 and Bhavya S. Doshi1,2.
1 Division of Hematology, Children's Hospital of Philadelphia, Philadelphia PA USA.
2 Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia PA USA.
3 Raymond G. Perelman Center for Cellular and Molecular Therapeutics, Children's Hospital of Philadelphia, Philadelphia PA USA.
Correspondence to: Valder R. Arruda, MD, PhD. Children’s
Hospital of Philadelphia, 3501 Civic Center Blvd, 5056 Colket
Translational Research Center, Philadelphia, PA 19104. Tel.: (215)
590-4907, Fax: (215) 590-3660. E-mail:
arruda@email.chop.edu Bhavya
S. Doshi, MD. Children’s Hospital of Philadelphia, 3501 Civic Center
Blvd, 5024 Colket Translational Research Center, Philadelphia, PA
19104. Tel.: (215) 590-3437, Fax: (215) 590-3992. E-mail:
doshibs@email.chop.edu
Published: September 1, 2020
Received: May 15, 2020
Accepted: August 19, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020069 DOI
10.4084/MJHID.2020.069
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Therapy
for hemophilia has evolved in the last 40 years from plasma-based
concentrates to recombinant proteins and, more recently, to non-factor
therapeutics. Along this same timeline, research in adeno-associated
viral (AAV) based gene therapy vectors has provided the framework for
early phase clinical trials initially for hemophilia B (HB) and now for
hemophilia A. Successive lessons learned from early HB trials have
paved the way for current advanced phase trials. Nevertheless,
questions linger regarding 1) the optimal balance of vector dose to
transgene expression, 2) amount and durability of transgene expression
required, and 3) long-term safety. Some trials have demonstrated unique
findings not seen previously regarding transient elevation of liver
enzymes, immunogenicity of the vector capsid, and loss of transgene
expression. This review will provide an update on the clinical AAV gene
therapy trials in hemophilia and address the questions above. A
thoughtful and rationally approached expansion of gene therapy to the
clinics would certainly be a welcome addition to the arsenal of options
for hemophilia therapy. Further, the global impact of gene therapy
could be vastly improved by expanding eligibility to different patient
populations and to developing nations. With the advances made to date,
it is possible to envision a shift from the early goal of simply
increasing life expectancy to a significant improvement in quality of
life by reduction in spontaneous bleeding episodes and disease
complications.
|
Introduction
Hemophilia is a bleeding disorder that results from mutations in the F8 or F9
genes encoding coagulation factors VIII (FVIII) or IX (FIX),
respectively. Deficiency or dysfunction of these clotting factors
disrupts the coagulation system and results in frequent, spontaneous
bleeding into the joints leading to chronic arthropathy, the hallmark
of severe disease (<1% normal FVIII or FIX activity). In severe
disease, people with hemophilia (PwH) are infused intravenously with
recombinant or plasma-derived factor concentrates for prophylaxis
against joint bleeds.[1] In patients with FVIII
deficiency or hemophilia A (HA), replacement therapy with standard
half-life products is required [2-3] times per week, whereas therapy
with standard half-life FIX in hemophilia B (HB) is required twice per
week. The advent of extended half-life (EHL) products[2,3] has
dramatically changed the infusion burden for HB patients to as
infrequently as once every 2 weeks, whereas currently licensed EHL
FVIII products have not had as dramatic of an increased half-life (up
to 1.5-fold) presumably due to the limitations imposed by von
Willebrand factor (VWF), the carrier for FVIII in circulation.[4]
Recent advances with non-factor therapies (NFTs) that either mimic the
cofactor function of activated FVIII (emicizumab)[5,6] or aim to
"rebalance" the coagulation system by decreasing natural anticoagulants
(antithrombin,[7] tissue factor pathway inhibitor,[8] and protein
C[9,10]) are revolutionizing the need for intravenous factor therapy
for prophylaxis but at this time cannot be used to treat bleeding
episodes (see the chapter by Dr. Makris and Dr. Castaman) and have been
associated with thrombotic complications in some patients.[11,12]
Robust
preclinical development of a liver-directed gene-based therapeutic
approach for hemophilia has culminated in promising clinical trial data
by several independent groups (Figure 1).
These trials support the prospect of a one-time infusion that could
modify the hemophilia phenotype, thus offering several potential
advantages compared to the current system of therapies. Hemophilia
served as a model disease for gene therapy trials due to its monogenic
nature, straightforward assessment of the efficacy by measurement of
circulating FVIII or FIX levels, and easily quantifiable clinical
endpoints such as bleeding rates and consumption of clotting factor
concentrates. Further supporting its appeal is the ability to improve
outcomes with even the modest efficacy of raising factor levels to
>1% (the goal of prophylactic factor replacement). First-in-human
gene therapy trials for hemophilia using retroviral or adenoviral
vector for liver gene therapy, or non-viral vector-based approaches for
skin fibroblast transduction and implantation into the omentum were
hampered by limited and transient efficacy and immune responses to some
viral vectors.[13] Moreover, the use of integrating murine retroviral
vector to genetically modified hematopoietic stem and progenitor cells
(HSPC) for some primary immunodeficiencies raised concerns for
oncogenicity.[14] The hemophilia clinical studies in progress are based
on the use of recombinant adeno-associated viral (rAAV) vectors, which
have demonstrated efficacy and safety. In some AAV-based strategies,
long-term improvement of the disease phenotype with an excellent safety
profile was achieved; these data will be discussed below. Strategies
using rAAV vectors targeting skeletal muscle for HB[15,16] or
lentiviral vectors for transduction of HSPC for HA[17] are being
planned or ongoing, respectively; these will not be discussed here.
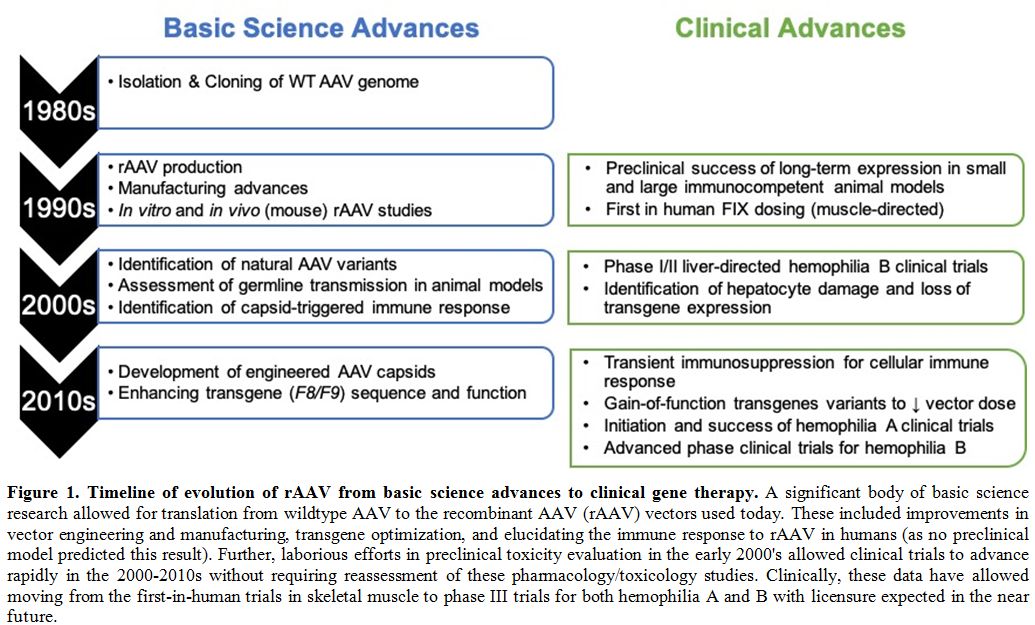 |
Figure 1. Timeline of evolution of rAAV from basic science advances to clinical gene therapy.
A significant body of basic science research allowed for translation
from wildtype AAV to the recombinant AAV (rAAV) vectors used today.
These included improvements in vector engineering and manufacturing,
transgene optimization, and elucidating the immune response to rAAV in
humans (as no preclinical model predicted this result). Further,
laborious efforts in preclinical toxicity evaluation in the early
2000's allowed clinical trials to advance rapidly in the 2000-2010s
without requiring reassessment of these pharmacology/toxicology
studies. Clinically, these data have allowed moving from the
first-in-human trials in skeletal muscle to phase III trials for both
hemophilia A and B with licensure expected in the near future.
|
AAV-Based Gene Therapy: the Facts
AAV
is a non-pathogenic, replication-deficient member of the parvovirus
family. Naturally occurring wildtype (WT) AAV consists of a
single-stranded DNA genome with two open reading frames flanked by
inverted tandem repeats (ITRs).[18] Binding of WT-AAV to heparan
sulfate proteoglycans on the host cell allows uptake and, replication
occurs following the entry into the host nucleus. Integration rates at
the AAVS1 site of chromosome 19 (which requires a functional rep
gene) vary from 45% in HeLa cells (aneuploid cells) to only 2.5% in
diploid human fibroblast cells.[19,20] In the rAAV vectors for
hemophilia applications, the WT-AAV coding sequences are replaced with
an F8 or F9
transgene under control of a tissue-specific promoter and flanked by
ITRs to allow packaging and production of the vector. Although the
genomes of the rAAV vectors remain largely episomal[21-24] as they lack
the rep gene, rAAV vectors
may integrate into the host DNA at other sites (reviewed in 24). Thus,
the integration pattern and its potential implication for oncogenesis
due to rAAV vectors differ from that of WT-AAV. Of the four currently
available distinct rAAV production platforms suitable for scaling up
vector production,[25] two have been most commonly used for hemophilia
gene therapy vectors. Hemophilia rAAV vectors to date have been
produced via either transfection of mammalian cells with naked plasmid
DNA or introduction of baculovirus expression vectors into Spodoptera frugiperda
(Sf9) insect cells followed by cell lysis and purification via cesium
chloride (CsCl) gradient sedimentation or ion exchange
chromatography.[26-30] The capsid-determined tropism of the various AAV
serotypes and promoter/enhancer elements used guide transgene
production to the target tissue of interest. To date, in hemophilia,
this has largely focused on liver-directed transgene expression under
the control of a liver-specific promoters/enhancers that restrict
expression to hepatocytes.[31,32]
Together, these distinct systems
and advances have facilitated the use of rAAV vectors for gene therapy
applications in hemophilia. Tables 1 and 2
summarize the multitude of rAAV-based gene therapy trials for
hemophilia A and B, respectively. These contemporary trials are the
product of decades of preclinical work and build on the successes and
lessons learned from the early gene therapy studies in HB.
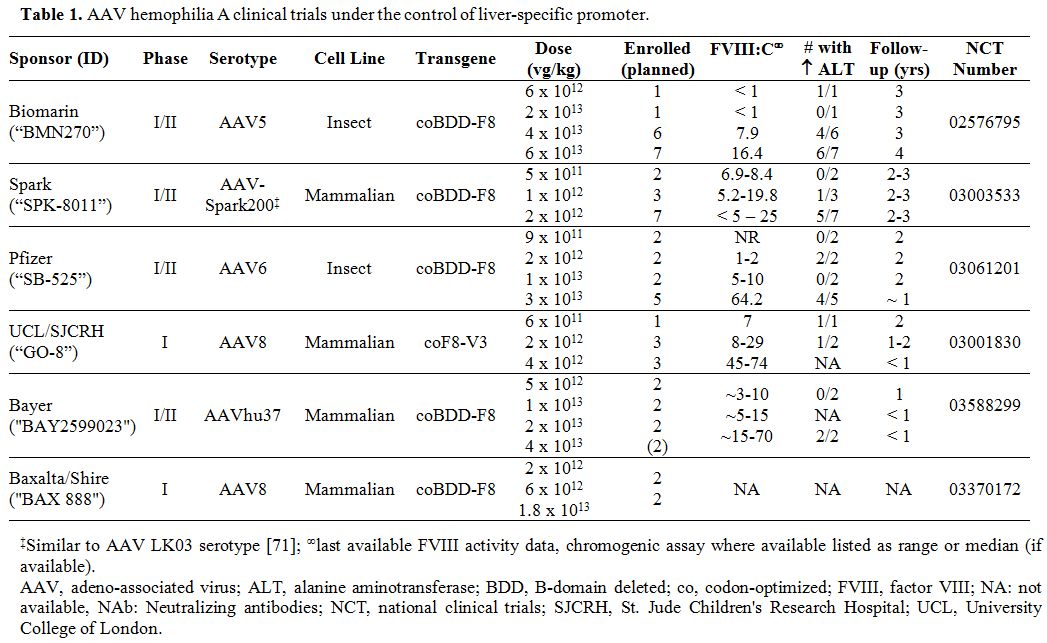 |
Table 1. AAV hemophilia A clinical trials under the control of liver-specific promoter |
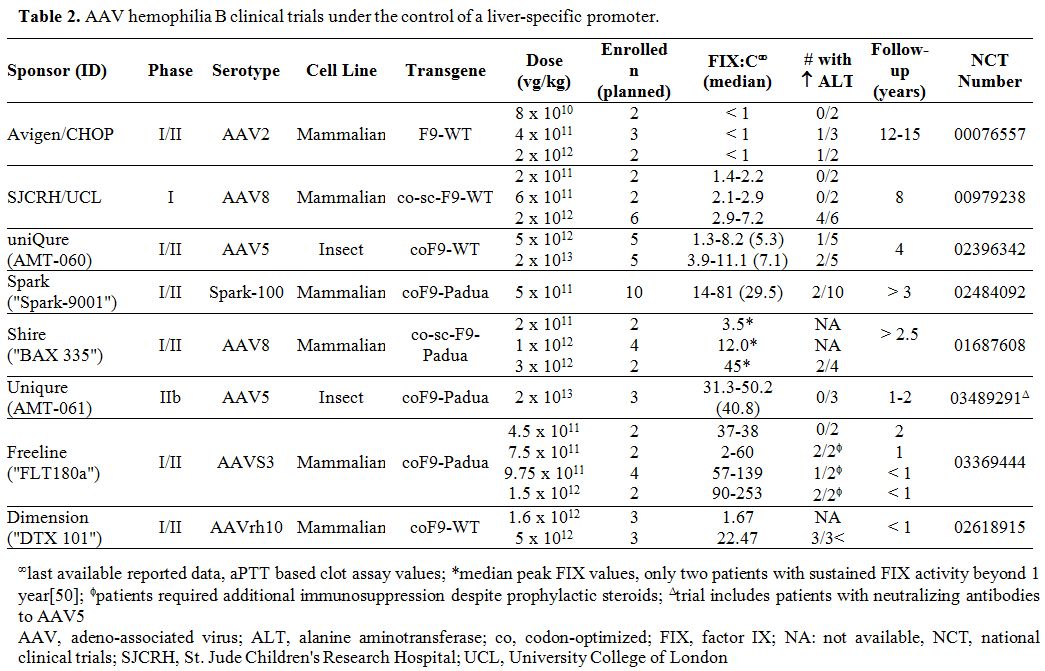 |
Table 2
AAV hemophilia B clinical trials under the control of a liver-specific promoter. |
AAV gene therapy for HB.
Due to the packaging constraints of the rAAV genome, the pioneering
rAAV hemophilia gene therapy studies were conducted in HB as the F9
cDNA is 1.6 kb in size.[33] The evolution of these early trials was
guided by both advances in the basic understanding of rAAV and
enhancements in vector production (Figure 1). The first-in-human rAAV F9 gene therapy trial utilized a ubiquitous cytomegalovirus (CMV) promoter/enhancer, and the rAAV2 serotype (rAAV2-CMV-F9-WT)
injected into skeletal muscle and demonstrated safe and prolonged
local, but not systemic, FIX expression.[34,35] This expression was not
hampered by either pre-existing neutralizing antibodies (NAbs) to rAAV2
or a post-infusion immune response to the vector and/or transgene. The
excellent short and long-term safety profile of this trial motivated
studies targeting the liver (as it is the natural site of FIX
production). The trial sponsored by Avigen and Children's Hospital of
Philadelphia (CHOP) of rAAV2-F9 under control of a liver-specific promoter (rAAV2-hAAT-F9-WT) administered the vector via the hepatic artery in 7 subjects (Table 2).[36] The low and mid-dose cohorts were intentionally subtherapeutic. In the high dose cohort (2x1012
vg/kg, n=2), pre-existing anti-rAAV2 NAbs did preclude transgene
expression in one subject, in contrast to the prior skeletal muscle
trial.[35] Another high-dose treated patient initially achieved a FIX
level of 11% but lost FIX activity concurrent with a transient rise in
alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
levels, markers of hepatocyte damage. After several attempts by many
groups to develop preclinical models to understand this phenomenon, it
became clear that such a complication is observed only in humans.
After consultation with regulatory agencies, another subject (Subject G) was dosed with subtherapeutic vector at 4 x 1011
vg/kg with a planned longitudinal collection of peripheral blood
mononuclear cells (PBMCs) to test for potential cellular immune
responses to the two neoantigens (rAAV capsid protein and FIX) using a
sensitive and specific technique, the interferon-γ
(IFNγ
)
enzyme-linked immunosorbent spot (ELISPOT). These studies suggested
that the underlying mechanism of this toxicity is likely a cytotoxic T
cell-mediated immune response against the vector capsid sequences
displayed on hepatocytes with resultant loss of transduced cells and
consequent constraint on transgene expression. There was no evidence of
cellular immune responses to FIX. There was, however, a temporal
association between the expansion of the rAAV2-capsid cellular
response, and a rise in the liver enzymes (ALT/AST).[37]
These
data guided the third defining HB trial, led by St. Jude Children's
Research Hospital (SJCRH) and University College of London (UCL), which
used rAAV8-LP1-F9-WT at escalating doses in 10 men with HB.[38,39] This study was the first to 1) infuse an F9
vector via a peripheral vein (which was made possible by the strong
liver tropism of AAV8 compared to AAV2) and 2) demonstrate that
initiating prednisone within 48 hours of noting a rise in ALT or drop
in FIX could limit the loss of FIX expression in vector-infused
patients. Further, this trial supported the dose-dependency of the
cellular immune response to the capsid as none of the patients in the
low (n=2) or intermediate (n=2) dose cohorts demonstrated an ALT rise (Table 2),
whereas 4 of 6 subjects (66%) in the high dose cohort showed evidence
of a cellular immune response. Over 8 years of follow-up, these
high-dose subjects have maintained FIX levels of 2.9-7.2% (Table 2).[40]
These
studies imparted two critical lessons. First, patients with
neutralizing antibodies against the AAV serotype should be excluded to
avoid the inhibitory effect on gene expression. Second, as there are no
biomarkers that predict the onset of rAAV capsid-mediated cellular
response, close monitoring of liver enzymes and factor levels should be
used as surrogate markers for the ongoing cellular responses in
real-time. Although ELISPOT assays are the most accurate for the
diagnosis of T cell responses, the turn-around time of the assay is not
ideal due to the need for rapid initiation of therapeutic intervention
to stop or control the loss of transduced cells and transgene
expression. Consequently, current clinical trials use the ALT as a
biomarker of potential capsid-directed cellular immune response and a
hallmark of liver damage; ALT is more sensitive to hepatocyte damage
than AST and has a longer half-life.[41]
These studies paved the way for the trial sponsored by Spark Therapeutics, which leveraged the hyperactive F9 variant, F9-Padua, with the goal to decrease the therapeutic vector dose while increasing transgene activity.[42] F9-Padua
results in an arginine to lysine substitution at position 338 in the
FIX protein and has a specific activity (activity to antigen ratio) of
> 8.[43] Thus, even small amounts of FIX antigen can provide
hemostatically normal FIX activity without increasing the risk of
thrombosis[44-46] compared to FIX-WT. A trial of 10 subjects injected
with 5 x 1011 vg/kg of rAAV-Spark100-F9-Padua
resulted in sustained FIX activity of ~30% of normal over multiple
years of follow-up and only 2/10 (20%) subjects required prednisone
therapy for a capsid-mediated immune response, which correlated with a
rise in ALT and decline in the FIX activity.[47] Despite decreasing the
vector dose 4-fold, a mean FIX activity of 30% was achieved, which is
~15-fold higher than with FIX-WT at a similar dose in the SJCRH trial (Figure 2).
It should be noted that in the Shire-sponsored rAAV8-F9-Padua trial,
loss of transgene expression in the high dose cohort (3 x 1012
vg/kg) could not be rescued in all patients,[48-50] which may be due to
higher vector doses and/or differences in vector content including CpG
islands.[51,52]
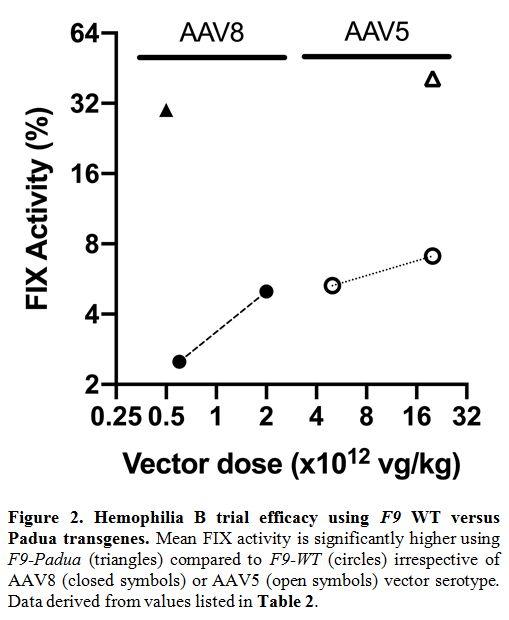 |
Figure 2. Hemophilia B trial efficacy using F9 WT versus Padua transgenes. Mean FIX activity is significantly higher using F9-Padua (triangles) compared to F9-WT (circles) irrespective of AAV8 (closed symbols) or AAV5 (open symbols) vector serotype. Data derived from values listed in Table 2..
|
Data from the Freeline sponsored trial using a novel serotype with liver tropism (AAVS3) encoding F9-Padua has recently been presented across four cohorts from 4.5 x 1011 to 1.5 x 1012
vg/kg. All patients received prophylactic steroids with varying
initiation times, depending on the cohort. In the lowest dose group,
there was no transaminitis, and FIX levels showed an increase while on
steroids with plateau levels at 2 years of 37-38%.[53,54] However, the
initial plan to scale up to 1.5 x 1012
vg/kg resulted in safety concerns such as transaminitis in both
subjects, which required methylprednisolone and tacrolimus to control.
One subject expressing supra-therapeutic FIX levels (peak 520%)
developed a local thrombotic complication at an arteriovenous fistula
when weaned off prophylactic anticoagulation.[55,56] Consequently, the
trial suspended this dose, and two additional intermediate-dose cohorts
were tested (Table 2). All
subjects received prophylactic steroid therapy, and tacrolimus was
added to control transaminitis either therapeutically or
prophylactically. Therapeutic levels of FIX were achieved in both
cohorts in a dose-dependent manner (the relatively short follow-up
period prevents firm efficacy conclusions at this point).
The uniQure-sponsored trials have utilized an Sf9 insect cell line and baculovirus production system to develop the rAAV5-LP1-F9
cDNA therapy platform encoding either FIX-WT (AMT-060)57 or FIX-Padua
(AMT-061).[58,59] A potential advantage of rAAV5 is the possibility of
a lower prevalence of NAbs in the general population compared to the
other AAV serotypes and/or lower avidity of anti-AAV5 NAbs.[60,61] It
should be noted that there is a wide range in the reported prevalence
of NAbs to AAV, including AAV5, in the general and hemophilia
populations due to variability in geographic origin of
populations[62-65] but also likely from differences in the assay
techniques. Data from uniQure's porphyria gene therapy trial of rAAV5
at 5 x 1011 – 1.8 x 1013
vg/kg demonstrated a lack of capsid-triggered immune response at those
doses; albeit none of the patients had therapeutic transgene levels
either.[66] In the AMT-06, 0 trial, 1/5 in the low dose, and 2/5 in the
high dose group (2 x 1013
vg/kg) had a transient rise in ALT (with onset ranging from 3 to 10
weeks). Still, there was no detectable capsid-mediated cellular immune
response as measured by IFN-γ
ELISPOT assay (the commonly used surrogate for cellular immunotoxicity)
or decrease in FIX levels.[57] The reason for this transient increase
in ALT remains unclear, although alcohol consumption and/or antibiotic
use were postulated as modulators, in part, of the liver damage. These
three patients received steroid therapy, but its therapeutic role is
not clear. The median FIX levels in the high dose cohort were 7.1%
compared to 5.3% in the low-dose cohort (4-fold lower dose) over 4
years of follow-up, suggesting a lack of clear linear dose-response (Figure 2). In the three patients treated with AMT-061 (encoding F9-Padua) at 2 x 1013
vg/kg, mean FIX activity is similar to the Spark trial with the same
transgene at about 40%,[58] but at 40-fold higher dose. As there was
seemingly no correlation between the presence of NAbs to rAAV5 and FIX
transgene expression in AMT-060,[61] candidates with anti-AAV5 NAbs
were not excluded from AMT-061, and the titers of the first three
patients are between 1:25 and 1:48.[59] However, this is a preliminary
finding, and further assessment of the detection of NAbs and the
ability of rAAV5 to overcome the presence of pre-existing NAbs is
necessary.
Early phase rAAV-based gene therapy for HB has
reached critical mass, providing a firm basis for current phase III
clinical trials. Collectively, FIX-Padua across distinct rAAV trials
has not shown increased immunogenicity or spontaneous thrombosis, which
is consistent with preclinical data in inhibitor-prone HB dogs.[45,46]
However, as seen in Figure 2,
there is a significant variation in the vector dose required to attain
similar FIX levels. Of note, the transgenes used in the rAAV5 trials
were also used in the rAAV2 and rAAV8 trials. Thus, F9 transgene is
unlikely to influence these discrepancies in therapeutic vector doses.
The differences in serotype, manufacturing process, post-translational
modification of the vector, or combination thereof may be responsible
for this discrepancy. A side-by-side comparison of these vector
production systems is highly desirable.
AAV Gene Therapy for HA. Although HA is more prevalent than HB, the generation of rAAV vectors that could efficiently accommodate the F8 cDNA (7 Kb) was challenging due to the limited capacity of the AAV genome at 4.7 kb. Modifications to the F8
transgene evolved over time. First, the B domain was truncated from
> 900 to 14 amino acids as it is not required for full procoagulant
activity of FVIII;[67] however, this was not sufficient to allow
cloning into rAAV vectors. Subsequently, a series of modifications in
the vector design, such as the generation of minimally sized effective
liver-specific promoters, enhancers, and other regulatory elements
allowed the generation of rAAV vectors that expressed FVIII. Finally,
the field developed codon-optimized (co) B-domain deleted (BDD)-F8
transgene(s) resulting in higher FVIII expression levels without
modifications to the amino acid sequence or need for additional space
in the transgene.
Several phase I and II HA trials are reporting therapeutic FVIII levels (Table 1)
in the moderate (1-5%) or mild (> 5%) hemophilia ranges. The vector
that is the furthest along the developmental pipeline for HA is
Biomarin's BMN-270, which is a rAAV5 vector carrying a co-BDD-F8
transgene.[68,69] In the high vector dose cohort (6 x 1013
vg/kg), median chromogenic FVIII levels were 55% (range 11-95%) in the
first year after treatment but, surprisingly, declined over the ensuing
four years to 16.4% (Figure 3A).[70]
Given that there was initially a dramatic increase in FVIII activities,
Biomarin also conducted a trial at an intermediate dose cohort (4 x 1013
vg/kg). The median chromogenic FVIII activity at year 1 for 5/6
subjects was 24% (one subject had levels < 3%) and over 3 years has
declined to 7.9% (Figure 3A).[70] The one-stage FVIII activity in these subjects was about 1.6-fold higher than their chromogenic activities (Figure 3B).
In the high dose cohort, following a rise in ALT in the first patient,
all subsequent subjects received prophylactic prednisone. Despite this,
all patients still developed a rise in ALT between weeks 3-28, and
there was no clear correlation between ALT improvement, FVIII activity,
and IFNγ
ELISPOT results.[69]
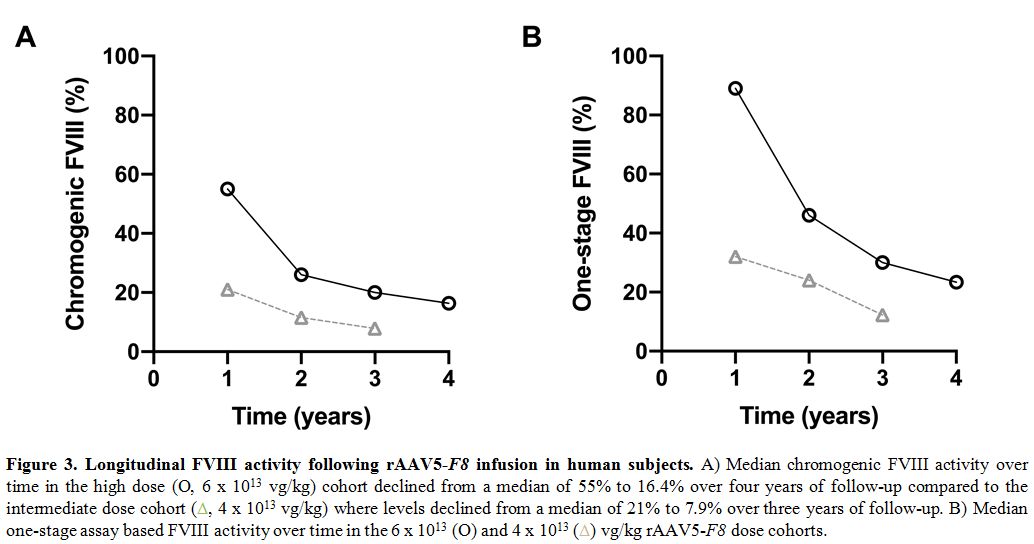 |
Figure 3. Longitudinal FVIII activity following rAAV5-F8 infusion in human subjects. A) Median chromogenic FVIII activity over time in the high dose (O, 6 x 1013
vg/kg) cohort declined from a median of 55% to 16.4% over four years of
follow-up compared to the intermediate dose cohort (triangles, 4 x 1013
vg/kg) where levels declined from a median of 21% to 7.9% over three
years of follow-up. B) Median one-stage assay based FVIII activity over
time in the 6 x 1013 (O) and 4 x 1013 (triangles) vg/kg rAAV5-F8 dose cohorts.
|
In the Spark Therapeutics trial of AAVSpark200-coBDD-F8 (similar to AAV serotype LK03)[71] within the high dose cohort (2 x 1012
vg/kg), five of seven subjects received steroids for either an increase
in ALT, declining FVIII levels, or a positive ELISPOT. Of these, two
experienced loss of FVIII activity that could not be rescued with
prednisone or methylprednisolone with levels falling below 5%.[72,73]
In contrast, one subject in the mid-dose cohort (1 x 1012
vg/kg) had a rise in ALT, and two of three had a decrease in FVIII
(treated with steroids) without an increase in ALT or positive IFNγ
ELISPOT. All three have maintained levels of ~5-20%. In the low dose
group, neither patient had a rise in ALT or loss of FVIII and have
maintained levels of ~7-8%. The UCL-SJCRH sponsored GO-8 trial (rAAV8-F8-V3), utilizing a hyperactive FVIII variant (F8-V3)
with amino acid insertions into the residual B domain sequence,[74]
demonstrated elevated ALT in two subjects at the low and mid-dose
cohorts (6 x 1011 and 2 x 1012 vg/kg, respectively) which resolved with prednisone but was not associated with a loss of FVIII activity.[75]
In
comparison to these mammalian cell line vectors, the Biomarin trial
utilizing an Sf9 insect cell line and baculovirus production
system-derived vector required a higher vector dose to achieve
therapeutic FVIII levels. For comparison, the low dose cohorts (6 x 1012 and 2 x 1013 vg/kg) in the Biomarin trial demonstrated < 1-2% FVIII activity (n = 1 per cohort) whereas a log-fold lower dose of 2 x 1012
vg/kg in the Spark and UCL/SJCRH trials did result in measurable FVIII
activity in the mild hemophilia range for those without an immune
response. In a recent publication, the authors argue that the initial
delay in achieving plateau FVIII activity levels with rAAV5-F8
is since the transgene is slightly larger than the vector's packaging
capacity and, as the positive and negative DNA strands of the transgene
are on separate virions, the full-length functional transcripts take
longer to assemble than with other trials,[68] but their findings do
not clearly support this. Further, preclinical studies in mouse and
canine models with rAAV2[76] or rAAV8[77] carrying the F8
gene split between two vectors (rAAV vectors carrying the light or
heavy chain) delivered simultaneously resulted in similar kinetics of
expression to single-chain FVIII. The unexpected loss of transgene
activity over time is also unusual in the context of rAAV liver gene
therapy. The authors argue that this is due to the turnover of
nucleated cells carrying stable full-length episomes, as measured by
sequencing analysis of PBMCs; again, these claims are highly
speculative at this point.[68]
The decline over time in FVIII levels seen in these subjects in the Biomarin trial has not been observed in the uniQure rAAV5-F9-WT
trial over 4-years of follow-up despite a 3-fold lower vector dose
compared to the Biomarin trial.[78] Further, the FIX expression in ten
men with HB injected with rAAV8-F9-WT
is stable over an extended period of ~ 8 years.[40] Thus, the
underlying mechanism of this loss of FVIII transgene expression remains
unclear; a combination of vector dose, vector manufacturing, and
transgene might impact the stability of the expression levels. At this
time, it is too early to identify the best performing rAAV system for
HA and long term follow up studies will be required to determine the
efficacy of any given strategy.
AAV-Based Gene Therapy: the Quandaries
These
significant advances in gene therapy for hemophilia make it likely to
enter the clinics in the coming years. There have been no sustained
adverse events documented in these trials with follow-up periods
ranging from < 3 years to more than 7 years with ongoing
observations. However, questions remain about (1) target factor level
and durability of response, (2) long-term follow-up requirements, (3)
the risk of genotoxicity, (4) expanding patient eligibility to
inhibitor patients and pediatric population, and (5) how to price and
pay for gene therapy.
1) Therapeutic transgene expression target and durability of response.
Initially, the goal in gene therapy trials was to bring factor levels
over the 1% necessary to convert a patient from severe to moderate
bleeding phenotype. In some subjects in the SJCRH/UCL and uniQure HB
trials, FIX levels of < 3% were not sufficient to prevent joint pain
and bleeds, and prophylaxis was necessary.[38,39,57] Further, higher
levels are likely required to prevent joint bleeds and stop
prophylaxis, as noted by a Dutch pediatric HA study that showed levels
> 12% were necessary to prevent joint bleeds.[79] A larger
U.S.-based study of adult and pediatric nonsevere HA and HB patients
estimated that FVIII or FIX levels > 20% of normal would be needed
to prevent hemarthrosis in individuals 25-44 years of age, the typical
age of subjects enrolled in early phase gene therapy trials.[80] On the
other hand, elevated FVIII (> 150 IU/dL) or FIX (> 129 IU/dL)
levels are associated with increased risk for thrombosis compared to
the general population.[81-85] Thus, true target FVIII or FIX activity
remains debatable.[86]
Understanding the optimal transgene level
is essential as the target FVIII or FIX level may affect the choice of
vector dose. Recent trials have used fixed doses for all enrolled
patients irrespective of underlying joint status or bleeding history.
Prior studies have noted variability in bleeding phenotype in patients
with severe hemophilia.[87,88] As vectors move from trials to clinical
practice, it may be important to consider these modifiers in choosing
the appropriate dose for each patient. The dose will need to be
carefully balanced against the risk of liver toxicity due to a cellular
immune response to the capsid (or unknown mechanisms).[89] At this
point, the dose-dependent cellular immune response to the vector capsid
does not correlate with the rise in ALT with insect cell-line derived
rAAV5 vectors. Whether this relationship will hold true in the ongoing
mammalian vector HA trials is unknown.
2) Long term follow-up requirements.
Gene therapy trials to date have typically been very selective in their
eligible population. For accurate assessments of both efficacy and
safety of a given strategy, long-term follow-up of these subjects is
necessary prior to and after approval of a product. Although the vector
infusion is given only once, patients will need to be followed for
recognition of potential unexpected findings, given the lack of
preclinical models that recapitulate the transient liver toxicity due
to cellular immune response to vector in humans. In contrast to the
loss of transgene activity in the BMN-270 trial, increasing gene
expression has been seen in long-term follow-up of a canine HA
model,[90] the reasons for these discrepancies continue to be
determined. Further, previous retroviral and AAV studies using a CMV
promoter in mice and dogs were complicated by gene silencing events of
transgene expression;[91,92] this has not been reported with the use of
liver-specific promoters.
In addition, adjustments of FVIII or
FIX levels may be necessary to accommodate for a subject's physical
activity and/or joint status (although vector re-administration at this
point is not feasible). Similarly, major trauma or surgery will also
require close monitoring and likely transient replacement therapy with
factor concentrates. The development of neutralizing alloantibodies
("inhibitors") to FVIII or FIX following gene therapy is an unlikely
scenario.[45,46,77,93,94] Still, little is known about subjects with
minimal exposure to factor concentrates prior to enrollment in rAAV
clinical trials wherein selected persons had > 20 exposure days.
Finally,
the risk of germline transmission of viral vectors is a major safety
concern. To date, gene therapy has been somatic in nature, and
preclinical studies were required by regulatory agencies prior to human
trials.[95,96] These did not show AAV in the semen of rabbits or dogs
receiving rAAV by intramuscular or portal vein injections,
respectively.[97] However, subjects in the rAAV2 trial did have
transient detection of vector sequences in the semen.[36,98,99]
Subsequent studies in rabbits using intravascular delivery of rAAV
vectors were associated with transient detection of a vector in semen
in a dose-dependent manner.[100,101] In addition, evidence supported
the concept that vector shedding into the semen did not require germ
cells, as the semen of vasectomized rabbits (i.e., lacking germ cells)
transiently contained vector sequences.[100] Although vector shedding
in the majority of the rAAV serotypes tested to date seem consistent
with these findings, the risk, if any, of inadvertent dissemination of
vector to germ cells needs to be determined after the development of
other natural or engineered rAAV vectors as these results may vary due
to vector and/or production platforms. The advice of the regulatory
agencies is to use barrier contraception while the semen contains rAAV
particles.[102]
3) Risk of genotoxicity.
In evaluating the safety in terms of potential genotoxicity due to rAAV
vectors, it is important to note that the recombinant AAV vector only
rarely integrates into the host DNA, whereas the WT-AAV may exhibit
latent infection via integration events mediated by the rep
gene. Thus, the rate and pattern of integration and risk of insertional
mutagenesis differ from the WT-AAV. Recombinant AAV vectors are poorly
integrating vectors with no preferential specific sites. For rAAV,
integration events, if any, occur at diverse locations depending on the
experimental model. Thus, findings from WT-AAV studies are not directly
relevant to rAAV vectors. Moreover, in some in vitro
experimental models, WT-AAV2 may, in fact, protect against tumor
formation.[103,104] Nevertheless, over the years, sporadic reports on
the risk of AAV integration and increased risk of tumor formation
raised safety concerns, particularly for genetic diseases with a long
life-expectancy such as hemophilia.
In 2001, animal studies
using rAAV in neonatal mice with MPS VII, with a high vector dose and a
strong enhancer element, demonstrated some integration, which led to
hepatocellular carcinoma (HCC).[105,106] Upon discussion with various
investigators and regulatory agencies, trials using rAAV vectors for
genetic disease were continued but with a commitment to long-term
follow up of the subjects who received a direct injection of the vector
for ~ 15 years. Some of these studies of rAAV liver gene therapy were
presented at scientific meetings, and early evaluation did not show
evidence of increased risk of tumor formation.[98] Subsequently, in
2007, a more detailed examination of the molecular evolution of the HCC
in neonatal mice showed that integration occurred largely at a miRNA
site or the Rian locus, which
is transcriptionally active in neonatal but not adult mice. This locus
is absent in vertebrates except for mice and rats.[104] Additional
studies in adult rodents, dogs, and non-human primates could not
confirm the increased risk of tumor formation by rAAV vectors.[107-112]
These risks can further be mitigated by modulating vector (dose,
promoter, enhancer) and subject (age, target tissue)
characteristics.[113] Interestingly, the vector constructs adapted for
several clinical studies for rAAV liver gene therapy seems to be
associated with the least, if any, risk of tumor formation in mouse
models.[113]
In 2015, Nault and colleagues showed the integration
pattern of WT-AAV in human subjects using a series of tissues from HCC
affected and normal areas.[114] In brief, only 11/193 samples showed
the integration of AAV in potential genes associated with HCC, but
samples were also positive for viral hepatitis and alcoholic liver
disease, and this study lacked data from healthy controls. These
findings were again informative of WT-AAV biology but are not
necessarily applicable for rAAV used in gene therapy.[103,115,116]
In
2017, Logan and colleagues found that an early rAAV2 vector retained a
small WT-AAV2 sequence in the 3' untranslated region (UTR) adjacent to
the ITR (derived from AAV2 and used in most rAAV constructs) which
contains a binding site of hepatic transcription factors (including
HNF1-α).[117]
This retained sequence can enhance transcription from the transgene
promoter in human hepatocytes and rodent livers. Further, this sequence
is captured within the 163-nucleotide frequent insertion region of the
WT-AAV2 genome that has been implicated in HCCs. However, there is no
definitive proof of insertional gene dysregulation by this sequence.
Emerging data presented only in abstract form of HA dogs injected with
rAAV carrying canine F8
showed integration events but without malignant transformation in
necropsy samples; the implications of these findings are still
unclear.[90] In addition, to date, numerous patients with hemophilia
have been treated with rAAV liver-directed gene therapy with no
reported significant safety or toxicity
concerns;[36,38,39,42,49,57,69,118] long-term data is still being
accumulated.
Overall, the likelihood of genotoxicity with rAAV
liver-directed gene therapy is likely low. It merits mention that in
the > 140 rAAV gene therapy trials targeting a variety of tissues,
none have reported oncogenesis. Although WT-AAV infection may be
associated with HCC, the risk of rAAV mediated HCC is currently
restricted to integration events into a murine-only genetic locus that
is active in neonatal mice. However, as shown by recent studies,
certain vectors may integrate into the genome indiscriminately. Of
note, the long-term follow-up of subjects from the CHOP/Avigen rAAV2-F9 trial at 12-15 years did not show evidence of tumors via measurement of tumor markers or liver enzymes.[98,119]
Current
guidelines from the Food and Drug Administration (FDA) and European
Medicines Agency (EMA) recommend a 5-year follow-up period for
non-integrating gene therapy vectors,[102,120] such as rAAV, as opposed
to prior guidelines which recommended 15 years. The rationale for this
shorter term of follow-up may no longer be applicable, especially given
the unprecedented fall in FVIII expression from the rAAV5-F8
trial over 4 years of ongoing observation. Further, the coupling in
recent trials of gene therapy with immunosuppressive regimens that
could modify safety and long-term complications also raises concerns.
Consequently, long-term clinical follow-up of patients from these early
trials should be undertaken to help inform the safety and efficacy of
liver-directed rAAV gene therapy in patients.
4) Expanding patient eligibility.
The next step in advancing gene therapy should be to allow for the
expansion of the target patient population to those who could benefit
from the recent advances in gene therapy. This includes patients
historically ineligible for gene therapy trials, including those with
inhibitors, patients < 18 years of age or who have fewer factor
exposure days, and those with NAbs to AAV serotypes.
4a) Patients with current or prior history of inhibitors to FVIII or FIX.
Due to the theoretical concern that gene therapy may increase the risk
of inhibitor formation, early phase clinical trials have excluded
patients with current inhibitors or a history of inhibitors to FVIII or
FIX. To date, data from both HA and HB subjects have not shown any
evidence of inhibitor development following gene therapy. AAV
liver-directed gene therapy in canine models of HA and HB demonstrate a
favorable bias towards inducing immune tolerance to canine FVIII or FIX
in inhibitor-prone HA[77] and HB dogs,[110,121]
respectively. Our laboratory has shown great promise for gene therapy
to provide the dual function of inducing immune tolerance to eradicate
inhibitors and provide lifelong endogenous prophylaxis in large animal
models of hemophilia A[93] and B[45] These preclinical studies suggest
that the liver-restricted endogenous expression of FVIII or FIX allows
for the induction of immune tolerance, at least in part in the HA
models, by the upregulation of a regulatory T cell pool.[93] Thus, the
possibility of using rAAV liver gene therapy for inhibitor eradication
would open a new therapeutic avenue to fulfill an unmet medical need.
Inhibitor eradication via immune tolerance induction (ITI) regimens is
costly, prone to catheter-related thrombotic and infectious
complications in pediatric patients,[122] and is successful in 60-70%
of patients with good prognostic risk.[123] In order to maintain
tolerance, modern-day practice is to continue factor prophylaxis 2-3
times per week. The use of emicizumab as a prophylactic hemostatic
regimen for inhibitor and non-inhibitor HA patients is now largely
accepted;[5,6] however, treatment of breakthrough bleeding episodes
still requires bypassing agents or FVIII therapy in these patients,
respectively. The use of prothrombin complex concentrates in some
patients has been associated with thrombosis.[11] However, the desired
outcome of standard ITI is the normalization of the hemostatic response
to FVIII to avoid bypassing agent therapy and this forms the motivation
for definitive inhibitor eradication in inhibitor patients; gene
therapy may hold a promising role in this context.[94] To date, one
clinical study has been planned to test gene therapy for inhibitor
eradication in HA; careful evaluation of preclinical studies in
relevant animal models will be critical to support such trials.
4b) Inclusion of pediatric hemophilia patients.
The pediatric patient population could also have tremendous benefit
from gene therapy approaches. Routine factor prophylaxis generally
requires indwelling central lines in infants and toddlers. The advent
of emicizumab and other NFTs, which are being developed to be
administered subcutaneously, could thus alleviate the burden in the
care of hemophilia. Moreover, EHL FIX products are highly effective in
HB patients with reduced frequency of injections,[124] but all of these
therapies would still require life-long infusions. Understanding the
durability of efficacy and risk of potential adverse events from the
different rAAV vector serotypes will allow consideration of the
inclusion of young patients in gene therapy trials.
4c) Inclusion of patients with NAbs to AAV capsid proteins.
Overall, 20-40% of candidates for intravascular AAV gene therapy are
not eligible due to the presence of NAbs to the vector capsid resulting
from cross-reactivity after natural exposure to WT-AAV.[125] To date,
it has been challenging to compare the efficacy of a given serotype
across distinct studies due to a lack of normalization of the assays as
most are developed "in house". Potential modifiers of the rates/titers
of NAbs could include (1) reporter gene, (2) assay technique whether
neutralizing assays, non-neutralizing antibody assays (ELISA) or
cellular assays, (3) presence or absence of empty vector capsids, (4)
age of the patient [65,126] and longitudinal studies in the same
population over time,[63] (5) underlying disease,[62,63,65,126] and (6)
vector manufacturing. Some candidates may also test positive for more
than one serotype at a time. Further, the amount of antigen (AAV capsid
protein) delivered by a gene therapy vector is likely to be orders of
magnitude higher than natural infection, and whether these immune
responses are similar or not remains to be determined. Finally,
although antibodies largely mediate inhibition of vector transduction
to the AAV capsid, there is evidence that, despite plasma depletion of
IgG, there may still be some inhibitory effect on the vector
transduction.
Several attempts aimed at the identification of
either naturally occurring or engineered AAV serotypes are in
development. However, to date, no candidate serotype allows for vector
administration without neutralizing effect or vector
re-administration.[127] The latter especially may be needed to rescue
transgene expression from either (a) the loss of the transduced cells
by liver toxicity or (b) dilution of the transgene expression levels
when delivered to young children, as the non-integrating nature of AAV
precludes effective transfer of the therapeutic genes to the liver
daughter cells upon cell division. To date, there is no clear evidence
that such a strategy is highly effective in both preclinical and
clinical models. Early efforts, including transient
immunosuppression[128] or altering ratio of empty "decoy"
capsids,[40,127] have not been successful, but there are some promising
strategies. The use of plasmapheresis,[129] catheter-guided perfusion
of the portal vein to flush out NAbs,[130] and more recently the use of
IgG degrading enzyme of Streptococcus pyogenes (IdeS)[131] or Streptococcus zooepidermicus
(IdeZ)[132] could reduce enough anti-AAV IgG to allow vector efficacy.
However, most of these strategies have been tried in the presence of
low-titer NAbs (< 1:20); some require invasive procedures, and may
have other sequelae such as opportunistic infections.[133] A recent
study, employing rapamycin nanoparticles in conjunction with the rAAV
vector, allowed for induction of tolerance to that vector serotype in
mice and non-human primates, which could permit re-administration.[134]
Vector re-administration was also possible in a limited study of IdeS
in NHP, but additional safety data regarding anti-IdeS antibody
development and the need for readministration is needed. The current
lack of sound safety data using any of these strategies in combination
with gene therapy precludes firm conclusion on their utility to
circumvent anti-AAV NAbs. Although there is some preclinical and
clinical evidence that NAbs to AAV5 may not prevent transgene
expression,[61] this requires further study as delineated above. Table 3
outlines a hypothetical strategy for the expansion of hemophilia gene
therapy moving forward, and the efficacy and safety outcomes that
warrant close monitoring as access is expanded.
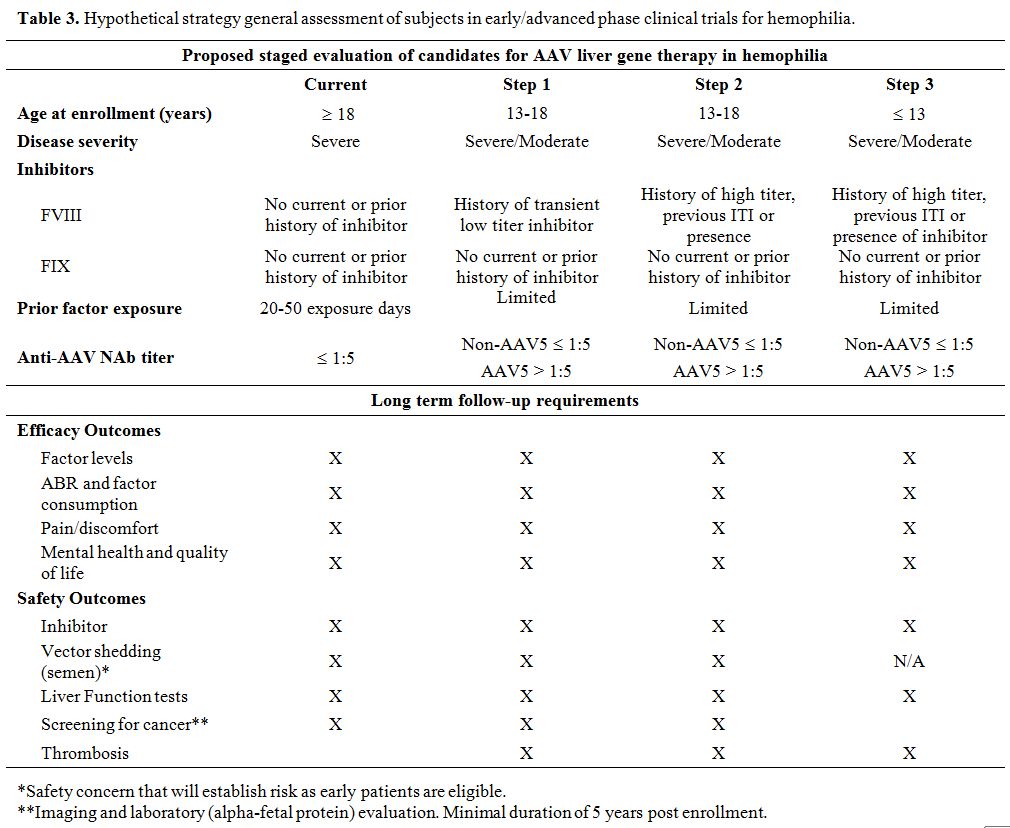 |
Table 3. Hypothetical strategy general assessment of subjects in early/advanced phase clinical trials for hemophilia.
|
I5) Price and reimbursement of gene therapy for hemophilia: challenges facing a "one and done" treatment for an orphan disease.
Motivated by the emerging success of rAAV liver gene therapy for
hemophilia, there is an ongoing debate regarding the cost and payment
for clinical gene therapy. A recent study analyzed the
cost-effectiveness of a given gene therapy approach compared to
standard, uncomplicated prophylaxis in non-inhibitor patients.[135] The
Markov Model used by Machin et al. assesses disease outcomes (bleeding,
surgical intervention, hospitalization) and quality of life against
cost in a hypothetical adult HA population on prophylaxis. In this
model, the cost of gene therapy was based on the first approved AAV
drug in the US, developed for inherited retinal degenerative disease
(estimated cost of $850,000). Interestingly, in hemophilia, the gene
therapy strategy was considered more cost-effective than protein-based
prophylaxis with a superior quality of life performance.
Both
payers and the hemophilia community are exploring ways to distribute
this cost either on a per annum basis (depending on durability) or with
a capped annuity. However, constraints in the existing healthcare
systems will need to be overcome to make this a reality. Further, the
initial production and development costs are driving current pricing;
the hope would be that as gene therapy matures in the clinics, these
costs will decline. When this occurs, gene therapy could become an
affordable and reasonable therapy in developing nations as well. Gene
therapy affords the enormous potential to alleviate the burden of
disease and improve the quality of life for people both in developed
and developing countries, but how gene therapy is priced will play a
significant role in its global impact.
Conclusions
A
copious body of preclinical and clinical work has brought hemophilia
gene therapy to the brink of becoming a tangible reality for
hemophilia. The phase I/II data show the ability to ameliorate the
bleeding phenotype and improve quality of life significantly and have
paved the way for phase III trials. Evaluating gene therapy in PwH with
pre-existing inhibitors or in pediatric subjects will likely be the
next frontier for rAAV. Together with NFTs, gene therapy-based
strategies point to a coming transformation in the treatment of
hemophilia.
These successes should not minimize the challenges
facing the gene therapy research community, including balancing vector
dose to limit the cellular immune response while maximizing therapeutic
efficacy and understanding the long-term risks from rAAV treatment.
Indeed, in most hemophilia trials, the risk of AAV capsid-triggered
cellular immune response and/or hepatotoxicity is proportional to the
vector dose. This dose-response toxicity should be taken into
consideration when assessing the development and choice of a
therapeutic approach for genetic diseases with a long life expectancy,
such as hemophilia. On the other hand, the dissociation between
elevated transaminases and cellular immune response to capsid in recent
trials is puzzling and deserves further study. Understanding this
finding will require cooperation between industry and academia as
differences in vector production, content, serotypes, and/or as yet
undisclosed modifications could help explain these discrepancies.
Higher vector doses likely carry a higher chance of inadvertent
long-term complications. For hemophilia, patients and providers need to
consider not only the goal FVIII or FIX expression level but also the
amount of vector required to achieve this goal.
In clinical
studies in HA, the considerable variability in the FVIII levels
following gene therapy and the loss of FVIII levels observed without
detection of AAV capsid-triggered immune response is a unique finding
with an undetermined mechanism, which might impact long-term efficacy.
It appears, at least, that this is not true for all rAAV vectors as
neither uniQure's rAAV5 nor the SJCRH rAAV8 HB trials that have
demonstrated a loss of FIX activity over 4 and 8 years of follow-up,
respectively.[40,78] Transduction and turnover of non-hepatocytes
likely do not contribute to this finding, given the use of a
liver-specific promoter. Transient expression outside the liver at very
early time points may take place,[136] but this is unlikely to affect
long-term persistence. The ability to re-administer vector might become
necessary if durable responses are not seen in the current trials.
While basic research progress is being made on this front as well, it
is not presently a reality in clinical practice.
Further, the
risks of integration with or without genotoxicity as well as germline
transmission for novel serotypes require further study with long-term
follow-up of subjects in the current trials to allow for safe and
rational expansion of eligible patient populations, specifically
pediatric patients. Finally, development for PwH residing in developing
nations should be considered to improve the global burden of
hemophilia. Despite several advances in therapeutics for hemophilia,
there has not been a significant decrease in the cost of care, which
places an undue disease burden on PwH in lower socioeconomic strata.
Making hemophilia gene therapy accessible and affordable for all will
require advocacy and innovative solutions from the entire community. As
the field moves forward and the above questions are answered, gene
therapy could undoubtedly be a welcomed therapeutic revolution and
provide health equity in hemophilia as proposed by Skinner et al.[137]
Acknowledgments
This
work was supported by grants from National Heart, Lung, and Blood
Institute grants U54- HL142012 (VRA) and RO1-HL137335-01 (VRA).
References
- Manco-Johnson, M.J., Abshire, T.C., Shapiro, A.D.,
Riske, B., Hacker, M.R., Kilcoyne, R., Ingram, J.D., Manco-Johnson,
M.L., Funk, S., Jacobson, L., Valentino, L.A., Hoots, W.K., Buchanan,
G.R., Dimichele, D., Recht, M., Brown, D., Leissinger, C., Bleak, S.,
Cohen, A., Mathew, P., Matsunaga, A., Medeiros, D., Nugent, D., Thomas,
G.A., Thompson, A.A., Mcredmond, K., Soucie, J.M., Austin, H., and
Evatt, B.L., Prophylaxis versus episodic treatment to prevent joint
disease in boys with severe hemophilia. N Engl J Med, 2007. 357(6): p.
535-544. https://doi.org/10.1056/NEJMoa067659 PMid:17687129
- Arruda,
V., Doshi, B., and Samelson-Jones, B., Novel approaches to hemophilia
therapy: successes and challenges. Blood, 2017. 130(21): p. 2251-2256.
https://doi.org/10.1182/blood-2017-08-742312 PMid:29018078
PMCid:PMC5813735
- Arruda, V.R., Doshi,
B.S., and Samelson-Jones, B.J., Emerging therapies for hemophilia:
controversies and unanswered questions. F1000Res, 2018. 7.
https://doi.org/10.12688/f1000research.12491.1 PMid:29770199
PMCid:PMC5931262
- Pipe, S.W., Montgomery,
R.R., Pratt, K.P., Lenting, P.J., and Lillicrap, D., Life in the shadow
of a dominant partner: the FVIII-VWF association and its clinical
implications for hemophilia A. Blood, 2016. 128(16): p. 2007-2016.
https://doi.org/10.1182/blood-2016-04-713289 PMid:27587878
PMCid:PMC5073181
- Mahlangu, J., Oldenburg,
J., Paz-Priel, I., Negrier, C., Niggli, M., Mancuso, M.E., Schmitt, C.,
Jiménez-Yuste, V., Kempton, C., Dhalluin, C., Callaghan, M.U., Bujan,
W., Shima, M., Adamkewicz, J.I., Asikanius, E., Levy, G.G., and
Kruse-Jarres, R., Emicizumab prophylaxis in patients who have
hemophilia A without inhibitors. New England Journal of Medicine, 2018.
379(9): p. 811-822. https://doi.org/10.1056/NEJMoa1803550 PMid:30157389
- Oldenburg,
J., Mahlangu, J.N., Kim, B., Schmitt, C., Callaghan, M.U., Young, G.,
Santagostino, E., Kruse-Jarres, R., Negrier, C., Kessler, C., Valente,
N., Asikanius, E., Levy, G.G., Windyga, J., and Shima, M., Emicizumab
prophylaxis in hemophilia A with inhibitors. New England Journal of
Medicine, 2017. 377(9): p. 809-818.
https://doi.org/10.1056/NEJMoa1703068 PMid:28691557
- Sehgal,
A., Barros, S., Ivanciu, L., Cooley, B., Qin, J., Racie, T., Hettinger,
J., Carioto, M., Jiang, Y., Brodsky, J., Prabhala, H., Zhang, X.,
Attarwala, H., Hutabarat, R., Foster, D., Milstein, S., Charisse, K.,
Kuchimanchi, S., Maier, M.A., Nechev, L., Kandasamy, P., Kel'in, A.V.,
Nair, J.K., Rajeev, K.G., Manoharan, M., Meyers, R., Sorensen, B.,
Simon, A.R., Dargaud, Y., Negrier, C., Camire, R.M., and Akinc, A., An
RNAi therapeutic targeting antithrombin to rebalance the coagulation
system and promote hemostasis in hemophilia. Nat Med, 2015. 21(5): p.
492-497. https://doi.org/10.1038/nm.3847 PMid:25849132
- Chowdary,
P., Lethagen, S., Friedrich, U., Brand, B., Hay, C., Karim, F.A.,
Klamroth, R., Knoebl, P., Laffan, M., Mahlangu, J., Miesbach, W.,
Nielsen, J.D., Martin-Salces, M., Angchaisuksiri, P., and The Explorer,
I., Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in
healthy volunteers and patients with hemophilia: a randomized first
human dose trial. J Thromb Haemost, 2015.
https://doi.org/10.1111/jth.12864 PMid:25641556
- Hamedani,
N.S., Ruhl, H., Zimmermann, J.J., Heiseler, T., Oldenburg, J., Mayer,
G., Potzsch, B., and Muller, J., In Vitro Evaluation of Aptamer-Based
Reversible Inhibition of Anticoagulant Activated Protein C as a Novel
Supportive Hemostatic Approach. Nucleic Acid Ther, 2016. 26(6): p.
355-362. https://doi.org/10.1089/nat.2016.0645 PMid:27736370
- Polderdijk,
S.G.I., Baglin, T.P., and Huntington, J.A., Targeting activated protein
C to treat hemophilia. Curr Opin Hematol, 2017. 24(5): p. 446-452.
https://doi.org/10.1097/MOH.0000000000000364 PMid:28632502
PMCid:PMC5548501
- Makris, M., Iorio, A.,
and Lenting, P.J., Emicizumab and thrombosis: The story so far. J
Thromb Haemost, 2019. 17(8): p. 1269-1272.
https://doi.org/10.1111/jth.14556 PMid:31368220
- Novo
Nordisk pauses the clinical trials investigating concizumab (anti-TFPI
mAB) in haemophilia A and B with or without inhibitors. 2020, Novo
Nordisk: Bagsværd, Denmark.
- Hough, C. and
Lillicrap, D., Gene therapy for hemophilia: an imperative to succeed. J
Thromb Haemost, 2005. 3(6): p. 1195-1205.
https://doi.org/10.1111/j.1538-7836.2005.01401.x PMid:15946210
- Hacein-Bey-Abina,
S., Von Kalle, C., Schmidt, M., Le Deist, F., Wulffraat, N., Mcintyre,
E., Radford, I., Villeval, J.L., Fraser, C.C., Cavazzana-Calvo, M., and
Fischer, A., A serious adverse event after successful gene therapy for
X-linked severe combined immunodeficiency. N Engl J Med, 2003. 348(3):
p. 255-256. https://doi.org/10.1056/NEJM200301163480314 PMid:12529469
- Arruda,
V.R., Stedman, H.H., Haurigot, V., Buchlis, G., Baila, S., Favaro, P.,
Chen, Y., Franck, H.G., Zhou, S., Wright, J.F., Couto, L.B., Jiang, H.,
Pierce, G.F., Bellinger, D.A., Mingozzi, F., Nichols, T.C., and High,
K.A., Peripheral transvenular delivery of adeno-associated viral
vectors to skeletal muscle as a novel therapy for hemophilia B. Blood,
2010. 115(23): p. 4678-4688.
https://doi.org/10.1182/blood-2009-12-261156 PMid:20335222
PMCid:PMC2890180
- Haurigot, V., Mingozzi,
F., Buchlis, G., Hui, D.J., Chen, Y., Basner-Tschakarjan, E., Arruda,
V.R., Radu, A., Franck, H.G., Wright, J.F., Zhou, S., Stedman, H.H.,
Bellinger, D.A., Nichols, T.C., and High, K.A., Safety of AAV factor IX
peripheral transvenular gene delivery to muscle in hemophilia B dogs.
Mol Ther, 2010. 18(7): p. 1318-1329. https://doi.org/10.1038/mt.2010.73
PMid:20424599 PMCid:PMC2911254
- Du,
L.M., Nurden, P., Nurden, A.T., Nichols, T.C., Bellinger, D.A., Jensen,
E.S., Haberichter, S.L., Merricks, E., Raymer, R.A., Fang, J.,
Koukouritaki, S.B., Jacobi, P.M., Hawkins, T.B., Cornetta, K., Shi, Q.,
and Wilcox, D.A., Platelet-targeted gene therapy with human factor VIII
establishes haemostasis in dogs with haemophilia A. Nat Commun, 2013.
4: p. 2773. https://doi.org/10.1038/ncomms3773 PMid:24253479
PMCid:PMC3868233
- Mezzina, M. and Merten,
O.W., Adeno-associated viruses. Methods Mol Biol, 2011. 737: p.
211-234. https://doi.org/10.1007/978-1-61779-095-9_9 PMid:21590399
- Huser,
D., Gogol-Doring, A., Chen, W., and Heilbronn, R., Adeno-associated
virus type 2 wildtype and vector-mediated genomic integration profiles
of human diploid fibroblasts analyzed by third-generation PacBio DNA
sequencing. J Virol, 2014. 88(19): p. 11253-11263.
https://doi.org/10.1128/JVI.01356-14 PMid:25031342 PMCid:PMC4178796
- Samulski,
R.J., Zhu, X., Xiao, X., Brook, J.D., Housman, D.E., Epstein, N., and
Hunter, L.A., Targeted integration of adeno-associated virus (AAV) into
human chromosome 19. EMBO J, 1991. 10(12): p. 3941-3950.
https://doi.org/10.1002/j.1460-2075.1991.tb04964.x PMid:1657596
PMCid:PMC453134
- Schnepp, B.C., Chulay,
J.D., Ye, G.J., Flotte, T.R., Trapnell, B.C., and Johnson, P.R.,
Recombinant Adeno-Associated Virus Vector Genomes Take the Form of
Long-Lived, Transcriptionally Competent Episomes in Human Muscle. Hum
Gene Ther, 2016. 27(1): p. 32-42. https://doi.org/10.1089/hum.2015.136
PMid:26650966 PMCid:PMC5374867
- Schnepp,
B.C., Clark, K.R., Klemanski, D.L., Pacak, C.A., and Johnson, P.R.,
Genetic fate of recombinant adeno-associated virus vector genomes in
muscle. J Virol, 2003. 77(6): p. 3495-3504.
https://doi.org/10.1128/JVI.77.6.3495-3504.2003 PMid:12610125
PMCid:PMC149530
- Song, S., Lu, Y., Choi,
Y.K., Han, Y., Tang, Q., Zhao, G., Berns, K.I., and Flotte, T.R.,
DNA-dependent PK inhibits adeno-associated virus DNA integration. Proc
Natl Acad Sci U S A, 2004. 101(7): p. 2112-2116.
https://doi.org/10.1073/pnas.0307833100 PMid:14766968 PMCid:PMC357060
- Berns,
K.I., The Unusual Properties of the AAV Inverted Terminal Repeat. Hum
Gene Ther, 2020. 31(9-10): p. 518-523.
https://doi.org/10.1089/hum.2020.017 PMid:32079423
- Smith, J.M., Grieger, J.C., and Samulski, R.J., Overcoming Bottlenecks in AAV Manufacturing for Gene Therapy.
- Grieger,
J.C., Soltys, S.M., and Samulski, R.J., Production of Recombinant
Adeno-associated Virus Vectors Using Suspension HEK293 Cells and
Continuous Harvest of Vector From the Culture Media for GMP FIX and
FLT1 Clinical Vector. Mol Ther, 2016. 24(2): p. 287-297.
https://doi.org/10.1038/mt.2015.187 PMid:26437810 PMCid:PMC4817810
- Matsushita,
T., Elliger, S., Elliger, C., Podsakoff, G., Villarreal, L., Kurtzman,
G.J., Iwaki, Y., and Colosi, P., Adeno-associated virus vectors can be
efficiently produced without helper virus. Gene Ther, 1998. 5(7): p.
938-945. https://doi.org/10.1038/sj.gt.3300680 PMid:9813665
- Kotin,
R.M., Large-scale recombinant adeno-associated virus production. Hum
Mol Genet, 2011. 20(R1): p. R2-6. https://doi.org/10.1093/hmg/ddr141
PMid:21531790 PMCid:PMC3095058
- i, C. and
Samulski, R.J., Engineering adeno-associated virus vectors for gene
therapy. Nat Rev Genet, 2020. 21(4): p. 255-272.
https://doi.org/10.1038/s41576-019-0205-4 PMid:32042148
- Robert,
M.A., Chahal, P.S., Audy, A., Kamen, A., Gilbert, R., and Gaillet, B.,
Manufacturing of recombinant adeno-associated viruses using mammalian
expression platforms. Biotechnol J, 2017. 12(3).
https://doi.org/10.1002/biot.201600193 PMid:28177193
- Xu,
L., Gao, C., Sands, M.S., Cai, S.R., Nichols, T.C., Bellinger, D.A.,
Raymer, R.A., Mccorquodale, S., and Ponder, K.P., Neonatal or
hepatocyte growth factor-potentiated adult gene therapy with a
retroviral vector results in therapeutic levels of canine factor IX for
hemophilia B. Blood, 2003. 101(10): p. 3924-3932.
https://doi.org/10.1182/blood-2002-10-3050 PMid:12531787
- Miao,
C.H., Ohashi, K., Patijn, G.A., Meuse, L., Ye, X., Thompson, A.R., and
Kay, M.A., Inclusion of the hepatic locus control region, an intron,
and untranslated region increases and stabilizes hepatic factor IX gene
expression in vivo but not in vitro. Mol Ther, 2000. 1(6): p. 522-532.
https://doi.org/10.1006/mthe.2000.0075 PMid:10933977
- Choo,
K.H., Gould, K.G., Rees, D.J., and Brownlee, G.G., Molecular cloning of
the gene for human anti-haemophilic factor IX. Nature, 1982. 299(5879):
p. 178-180. https://doi.org/10.1038/299178a0 PMid:6287289
- Buchlis,
G., Podsakoff, G.M., Radu, A., Hawk, S.M., Flake, A.W., Mingozzi, F.,
and High, K.A., Factor IX expression in skeletal muscle of a severe
hemophilia B patient 10 years after AAV-mediated gene transfer. Blood,
2012. 119(13): p. 3038-3041.
https://doi.org/10.1182/blood-2011-09-382317 PMid:22271447
PMCid:PMC3321866
- Manno, C.S., Chew, A.J.,
Hutchison, S., Larson, P.J., Herzog, R.W., Arruda, V.R., Tai, S.J.,
Ragni, M.V., Thompson, A., Ozelo, M., Couto, L.B., Leonard, D.G.,
Johnson, F.A., Mcclelland, A., Scallan, C., Skarsgard, E., Flake, A.W.,
Kay, M.A., High, K.A., and Glader, B., AAV-mediated factor IX gene
transfer to skeletal muscle in patients with severe hemophilia B.
Blood, 2003. 101(8): p. 2963-2972.
https://doi.org/10.1182/blood-2002-10-3296 PMid:12515715
- Manno,
C.S., Pierce, G.F., Arruda, V.R., Glader, B., Ragni, M., Rasko, J.J.,
Ozelo, M.C., Hoots, K., Blatt, P., Konkle, B., Dake, M., Kaye, R.,
Razavi, M., Zajko, A., Zehnder, J., Rustagi, P.K., Nakai, H., Chew, A.,
Leonard, D., Wright, J.F., Lessard, R.R., Sommer, J.M., Tigges, M.,
Sabatino, D., Luk, A., Jiang, H., Mingozzi, F., Couto, L., Ertl, H.C.,
High, K.A., and Kay, M.A., Successful transduction of liver in
hemophilia by AAV-Factor IX and limitations imposed by the host immune
response. Nat Med, 2006. 12(3): p. 342-347.
https://doi.org/10.1038/nm1358 PMid:16474400
- Mingozzi,
F., Maus, M.V., Hui, D.J., Sabatino, D.E., Murphy, S.L., Rasko, J.E.,
Ragni, M.V., Manno, C.S., Sommer, J., Jiang, H., Pierce, G.F., Ertl,
H.C., and High, K.A., CD8(+) T-cell responses to adeno-associated virus
capsid in humans. Nat Med, 2007. 13(4): p. 419-422.
https://doi.org/10.1038/nm1549 PMid:17369837
- Nathwani,
A.C., Reiss, U.M., Tuddenham, E.G., Rosales, C., Chowdary, P.,
Mcintosh, J., Della Peruta, M., Lheriteau, E., Patel, N., Raj, D.,
Riddell, A., Pie, J., Rangarajan, S., Bevan, D., Recht, M., Shen, Y.M.,
Halka, K.G., Basner-Tschakarjan, E., Mingozzi, F., High, K.A., Allay,
J., Kay, M.A., Ng, C.Y., Zhou, J., Cancio, M., Morton, C.L., Gray,
J.T., Srivastava, D., Nienhuis, A.W., and Davidoff, A.M., Long-term
safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J
Med, 2014. 371(21): p. 1994-2004. https://doi.org/10.1056/NEJMoa1407309
PMid:25409372 PMCid:PMC4278802
- Nathwani,
A.C., Tuddenham, E.G., Rangarajan, S., Rosales, C., Mcintosh, J.,
Linch, D.C., Chowdary, P., Riddell, A., Pie, A.J., Harrington, C.,
O'beirne, J., Smith, K., Pasi, J., Glader, B., Rustagi, P., Ng, C.Y.,
Kay, M.A., Zhou, J., Spence, Y., Morton, C.L., Allay, J., Coleman, J.,
Sleep, S., Cunningham, J.M., Srivastava, D., Basner-Tschakarjan, E.,
Mingozzi, F., High, K.A., Gray, J.T., Reiss, U.M., Nienhuis, A.W., and
Davidoff, A.M., Adenovirus-associated virus vector-mediated gene
transfer in hemophilia B. N Engl J Med, 2011. 365(25): p. 2357-2365.
https://doi.org/10.1056/NEJMoa1108046 PMid:22149959 PMCid:PMC3265081
- Nathwani,
A.C., Reiss, U., Tuddenham, E., Chowdary, P., Mcintosh, J., Riddell,
A., Pie, J., Mahlangu, J.N., Recht, M., and Shen, Y.-M.,
Adeno-Associated Mediated Gene Transfer for Hemophilia B: 8 Year Follow
up and Impact of Removing" Empty Viral Particles" on Safety and
Efficacy of Gene Transfer. 2018, Am Soc Hematology.
https://doi.org/10.1182/blood-2018-99-118334
- Moriles, K.E. and Azer, S.A., Alanine Amino Transferase (ALT), in StatPearls. 2020: Treasure Island (FL).
- George,
L.A., Sullivan, S.K., Giermasz, A., Rasko, J.E.J., Samelson-Jones,
B.J., Ducore, J., Cuker, A., Sullivan, L.M., Majumdar, S., Teitel, J.,
Mcguinn, C.E., Ragni, M.V., Luk, A.Y., Hui, D., Wright, J.F., Chen, Y.,
Liu, Y., Wachtel, K., Winters, A., Tiefenbacher, S., Arruda, V.R., Van
Der Loo, J.C.M., Zelenaia, O., Takefman, D., Carr, M.E., Couto, L.B.,
Anguela, X.M., and High, K.A., Hemophilia B Gene Therapy with a
High-Specific-Activity Factor IX Variant. N Engl J Med, 2017. 377(23):
p. 2215-2227. https://doi.org/10.1056/NEJMoa1708538 PMid:29211678
PMCid:PMC6029626
- Simioni, P., Tormene,
D., Tognin, G., Gavasso, S., Bulato, C., Iacobelli, N.P., Finn, J.D.,
Spiezia, L., Radu, C., and Arruda, V.R., X-linked thrombophilia with a
mutant factor IX (factor IX Padua). N Engl J Med, 2009. 361(17): p.
1671-1675. https://doi.org/10.1056/NEJMoa0904377 PMid:19846852
- Arruda,
V. and Samelson-Jones, B., Factor IX Padua: From Biochemistry to Gene
Therapy. Blood, 2016. 128(22): p. SCI-9.
https://doi.org/10.1182/blood.V128.22.SCI-9.SCI-9
- Crudele,
J.M., Finn, J.D., Siner, J.I., Martin, N.B., Niemeyer, G.P., Zhou, S.,
Mingozzi, F., Lothrop, C.D., Jr., and Arruda, V.R., AAV liver
expression of FIX-Padua prevents and eradicates FIX inhibitor without
increasing thrombogenicity in hemophilia B dogs and mice. Blood, 2015.
125(10): p. 1553-1561. https://doi.org/10.1182/blood-2014-07-588194
PMid:25568350 PMCid:PMC4351503
- Finn,
J.D., Nichols, T.C., Svoronos, N., Merricks, E.P., Bellenger, D.A.,
Zhou, S., Simioni, P., High, K.A., and Arruda, V.R., The efficacy and
the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs
treated by AAV muscle gene therapy. Blood, 2012. 120(23): p. 4521-4523.
https://doi.org/10.1182/blood-2012-06-440123 PMid:22919027
PMCid:PMC3512231
- George, L.A., Sullivan,
S.K., Rasko, J.E., Giermasz, A., Samelson-Jones, B.J., Ducore, J.M.,
Teitel, J.M., Mcguinn, C.E., Runowski, A.R., and Wright, F., Efficacy
and safety in 15 hemophilia B patients treated with the AAV gene
therapy vector Fidanacogene Elaparvovec and followed for at least 1
year. 2019, American Society of Hematology Washington, DC.
https://doi.org/10.1182/blood-2019-124091
- Monahan,
P.E., Sun, J., Gui, T., Hu, G., Hannah, W.B., Wichlan, D.G., Wu, Z.,
Grieger, J.C., Li, C., Suwanmanee, T., Stafford, D.W., Booth, C.J.,
Samulski, J.J., Kafri, T., Mcphee, S.W., and Samulski, R.J., Employing
a gain-of-function factor IX variant R338L to advance the efficacy and
safety of hemophilia B human gene therapy: preclinical evaluation
supporting an ongoing adeno-associated virus clinical trial. Hum Gene
Ther, 2015. 26(2): p. 69-81. https://doi.org/10.1089/hum.2014.106
PMid:25419787 PMCid:PMC4326268
- Monahan,
P.E., Walsh, C.E., Powell, J.S., Konkle, B., Josephson, N.C., Escobar,
M.A., Mcphee, S.J., Litchev, B., Cecerle, M., Ewenstein, B.M.,
Rottensteiner, H., Rose, M.D.I., Reipert, B.M., Samulski, R.J., Orloff,
J., and Scheiflinger, F., Update on phase 1/2 open-label trial of
BAX335, an adeno-associated virus 8 (AAV8) vector-based gene therapy
for program for phemophilia B. Journal of Thrombosis and Haemostasis,
2015. 13(Supplement 2): p. 87.
- Chapin,
J., Rottensteiner, H., Scheiflinger, F., and Monahan, P., An analysis
of bleeding rates and factor IX consumption in the phase I/II BAX 335
gene therapy trial in subjects with hemophilia B. Res Pract Thromb
Haemost, 2017. 1(Suppl. 1): p. 144.
- Pierce,
G. and Iorio, A., Past, present and future of haemophilia gene therapy:
From vectors and transgenes to known and unknown outcomes. Haemophilia,
2018. 24: p. 60-67. https://doi.org/10.1111/hae.13489 PMid:29878660
- Wright,
J.F., Codon Modification and PAMPs in Clinical AAV Vectors: The
Tortoise or the Hare? Mol Ther, 2020. 28(3): p. 701-703.
https://doi.org/10.1016/j.ymthe.2020.01.026 PMid:32035026
- Chowdary,
P., Shapiro, S., Davidoff, A.M., Reiss, U., Alade, R., Brooks, G.,
Dane, A., Mcintosh, J., Short, G., and Tuddenham, E., A single
intravenous infusion of FLT180a results in factor IX activity levels of
more than 40% and has the potential to provide a functional cure for
patients with haemophilia B. Blood, 2018. 132(Supplement 1): p.
631-631. https://doi.org/10.1182/blood-2018-99-118050
- Chowdary
P, S.S., Makris M, Evans G, Boyce S, Talks K, Dolan G, Reiss U,
Phillips M, Riddell a, Peralta Mr, Quaye M, Tuddenham E, Krop J, Short
G, Kar S, Smith a, Nathwani A. , A Novel Adeno Associated Virus (AAV)
Gene Therapy (FLT180a) Achieves Normal FIX Activity Levels in Severe
Hemophilia B (HB) Patients (B-AMAZE Study). Res Pract Thromb Haemost.,
2020. 4
- Carvalho, J., #ISTH2020 - FLT180a
Gene Therapy Shows Promise for Hemophilia B Patients in Phase 1/2
Trial, in Hemophilia News Today. 2020: Pensacola, FL.
- Isth,
A Novel Adeno Associated Virus (AAV) Gene Therapy (FLT180a) Achieves
Normal FIX Activity Levels in Severe Hemophilia B (HB) Patients
(B-AMAZE Study), in ISTH Congress Daily News. 2020.
- Miesbach,
W., Meijer, K., Coppens, M., Kampmann, P., Klamroth, R., Schutgens, R.,
Tangelder, M., Castaman, G., Schwable, J., Bonig, H., Seifried, E.,
Cattaneo, F., Meyer, C., and Leebeek, F.W.G., Gene therapy with
adeno-associated virus vector 5-human factor IX in adults with
hemophilia B. Blood, 2017. https://doi.org/10.1182/blood-2017-09-804419
PMid:29246900 PMCid:PMC5833265
- Pipe, S.,
Giermasz, A., Castaman, G., Key, N.S., Lattimore, S.U., Leebeek, F.,
Miesbach, W., Recht, M., Gomez, E., and Long, A., One Year Data from a
Phase 2b Trial of AMT-061 (AAV5-Padua hFIX variant), an Enhanced Vector
for Gene Transfer in Adults with Severe or Moderate-Severe Hemophilia
B. 2019, American Society of Hematology Washington, DC.
https://doi.org/10.1182/blood-2019-128765
- Pipe,
S.W., Giermasz, A., Castaman, G., Key, N., Lattimore, S.U., Leebeek,
F.W., Miesbach, W., Recht, M., Long, A., Gut, R., and Von Drygalski,
A., AMT-061 (AAV5-Padua hFIX variant) an enchanced vector for gene
transfer in adults with severe or moderate-severe hemophilia B:
follow-up up to 9 months in a phase 2b trial, in 71st Annual National
Hemophilia Foundation Bleeding Disorders Conference. 2019: Anaheim, CA.
- Boutin,
S., Monteilhet, V., Veron, P., Leborgne, C., Benveniste, O., Montus,
M.F., and Masurier, C., Prevalence of serum IgG and neutralizing
factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9
in the healthy population: implications for gene therapy using AAV
vectors. Hum Gene Ther, 2010. 21(6): p. 704-712.
https://doi.org/10.1089/hum.2009.182 PMid:20095819
- Majowicz,
A., Nijmeijer, B., Lampen, M.H., Spronck, L., De Haan, M., Petry, H.,
Van Deventer, S.J., Meyer, C., Tangelder, M., and Ferreira, V.,
Therapeutic hFIX Activity Achieved after Single AAV5-hFIX Treatment in
Hemophilia B Patients and NHPs with Pre-existing Anti-AAV5 NABs. Mol
Ther Methods Clin Dev, 2019. 14: p. 27-36.
https://doi.org/10.1016/j.omtm.2019.05.009 PMid:31276009
PMCid:PMC6586596
- Harrington, E.A., Sloan,
J.L., Manoli, I., Chandler, R.J., Schneider, M., Mcguire, P.J.,
Calcedo, R., Wilson, J.M., and Venditti, C.P., Neutralizing Antibodies
Against Adeno-Associated Viral Capsids in Patients with mut
Methylmalonic Acidemia. Hum Gene Ther, 2016. 27(5): p. 345-353.
https://doi.org/10.1089/hum.2015.092 PMid:26790480 PMCid:PMC4841085
- Li,
C., Narkbunnam, N., Samulski, R.J., Asokan, A., Hu, G., Jacobson, L.J.,
Manco-Johnson, M.J., Monahan, P.E., and Joint Outcome Study, I.,
Neutralizing antibodies against adeno-associated virus examined
prospectively in pediatric patients with hemophilia. Gene Ther, 2012.
19(3): p. 288-294. https://doi.org/10.1038/gt.2011.90 PMid:21697954
- Calcedo,
R., Vandenberghe, L.H., Gao, G., Lin, J., and Wilson, J.M., Worldwide
epidemiology of neutralizing antibodies to adeno-associated viruses. J
Infect Dis, 2009. 199(3): p. 381-390. https://doi.org/10.1086/595830
PMid:19133809
- Halbert, C.L., Miller,
A.D., Mcnamara, S., Emerson, J., Gibson, R.L., Ramsey, B., and Aitken,
M.L., Prevalence of neutralizing antibodies against adeno-associated
virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal
populations: Implications for gene therapy using AAV vectors. Hum Gene
Ther, 2006. 17(4): p. 440-447. https://doi.org/10.1089/hum.2006.17.440
PMid:16610931 PMCid:PMC4292890
- D'avola,
D., Lopez-Franco, E., Sangro, B., Paneda, A., Grossios, N., Gil-Farina,
I., Benito, A., Twisk, J., Paz, M., Ruiz, J., Schmidt, M., Petry, H.,
Harper, P., De Salamanca, R.E., Fontanellas, A., Prieto, J., and
Gonzalez-Aseguinolaza, G., Phase I open label liver-directed gene
therapy clinical trial for acute intermittent porphyria. J Hepatol,
2016. 65(4): p. 776-783. https://doi.org/10.1016/j.jhep.2016.05.012
PMid:27212246
- Lind, P., Larsson, K.,
Spira, J., Sydow-Backman, M., Almstedt, A., Gray, E., and Sandberg, H.,
Novel forms of B-domain deleted recombinant factor VIII molecules.
Construction and biochemical characterization. European Journal of
Biochemistry, 1995. 232: p. 19-27.
https://doi.org/10.1111/j.1432-1033.1995.tb20776.x PMid:7556150
- Pasi,
K.J., Rangarajan, S., Mitchell, N., Lester, W., Symington, E., Madan,
B., Laffan, M., Russell, C.B., Li, M., Pierce, G.F., and Wong, W.Y.,
Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N
Engl J Med, 2020. 382(1): p. 29-40.
https://doi.org/10.1056/NEJMoa1908490 PMid:31893514
- Rangarajan,
S., Walsh, L., Lester, W., Perry, D., Madan, B., Laffan, M., Yu, H.,
Vettermann, C., Pierce, G.F., Wong, W.Y., and Pasi, K.J., AAV5-Factor
VIII Gene Transfer in Severe Hemophilia A. N Engl J Med, 2017. 377(26):
p. 2519-2530. https://doi.org/10.1056/NEJMoa1708483 PMid:29224506
- Biomarin,
BioMarin Provides Additional Data from Recent 4 Year Update of Ongoing
Phase 1/2 Study of Valoctocogene Roxaparvovec Gene Therapy for Severe
Hemophilia A in Late-Breaking Oral Presentation at World Federation of
Hemophilia Virtual Summit. 2020: San Rafael, CA.
- Lisowski,
L., Dane, A.P., Chu, K., Zhang, Y., Cunningham, S.C., Wilson, E.M.,
Nygaard, S., Grompe, M., Alexander, I.E., and Kay, M.A., Selection and
evaluation of clinically relevant AAV variants in a xenograft liver
model. Nature, 2014. 506(7488): p. 382-386.
https://doi.org/10.1038/nature12875 PMid:24390344 PMCid:PMC3939040
- High,
K.A., George, L.A., Eyster, M.E., Sullivan, S.K., Ragni, M.V., Croteau,
S.E., Samelson-Jones, B.J., Evans, M., Joseney-Antoine, M., and
Macdougall, A., A phase 1/2 trial of investigational SPK-8011 in
hemophilia A demonstrates durable expression and prevention of bleeds.
Blood, 2018. 132(Supplement 1): p. 487-487.
https://doi.org/10.1182/blood-2018-99-115495
- George
L, Eyster E, Ragni M, Sullivan S, Samelson-Jones B, Evans M, Macdougall
A, Curran M, Tompkins S, Wachtel K, Takefman D, Reape K, Mingozzi F,
Monahan P, Anguela X, and K., H., Phase I/II Trial of SPK-8011: Stable
and Durable FVIII Expression for >2 Years with Significant ABR
Improvements in Initial Dose Cohorts Following AAV-Mediated FVIII Gene
Transfer for Hemophilia A [abstract]. . Res Pract Thromb Haemost, 2020.
4.
- Mcintosh, J., Lenting, P.J., Rosales,
C., Lee, D., Rabbanian, S., Raj, D., Patel, N., Tuddenham, E.G.,
Christophe, O.D., Mcvey, J.H., Waddington, S., Nienhuis, A.W., Gray,
J.T., Fagone, P., Mingozzi, F., Zhou, S.Z., High, K.A., Cancio, M., Ng,
C.Y., Zhou, J., Morton, C.L., Davidoff, A.M., and Nathwani, A.C.,
Therapeutic levels of FVIII following a single peripheral vein
administration of rAAV vector encoding a novel human factor VIII
variant. Blood, 2013. 121(17): p. 3335-3344.
https://doi.org/10.1182/blood-2012-10-462200 PMid:23426947
PMCid:PMC3637010
- Nathwani, A.C.,
Tuddenham, E., Chowdary, P., Mcintosh, J., Lee, D., Rosales, C.,
Phillips, M., Pie, J., Junfang, Z., and Meagher, M.M., GO-8:
preliminary results of a phase I/II dose escalation trial of gene
therapy for haemophilia a using a novel human factor VIII variant.
Blood, 2018. 132(Supplement 1): p. 489-489.
https://doi.org/10.1182/blood-2018-99-118256
- Mah,
C., Sarkar, R., Zolotukhin, I., Schleissing, M., Xiao, X., Kazazian,
H.H., and Byrne, B.J., Dual vectors expressing murine factor VIII
result in sustained correction of hemophilia A mice. Hum Gene Ther,
2003. 14(2): p. 143-152. https://doi.org/10.1089/104303403321070838
PMid:12614565
- Sabatino, D.E., Lange,
A.M., Altynova, E.S., Sarkar, R., Zhou, S., Merricks, E.P., Franck,
H.G., Nichols, T.C., Arruda, V.R., and Kazazian, H.H., Jr., Efficacy
and safety of long-term prophylaxis in severe hemophilia A dogs
following liver gene therapy using AAV vectors. Mol Ther, 2011. 19(3):
p. 442-449. https://doi.org/10.1038/mt.2010.240 PMid:21081906
PMCid:PMC3048175
- Miesbach, W., Meijer,
K., Coppens, M., Kampmann, P., Klamroth, D., Schutgens, R., Castaman,
G., Seifried, E., Schwaeble, J., Bönig, H., Sawyer, E.K., and Leebeek,
F.W.G., Stable FIX Expression and Durable Reductions in Bleeding and
Factor IX Consumption for up to 4 Years Following AMT-060 Gene Therapy
in Adults with Severe or Moderate-Severe Hemophilia B. Blood, 2019.
134(Supplement_1): p. 2059-2059.
https://doi.org/10.1182/blood-2019-122535
- Den
Uijl, I.E., Mauser Bunschoten, E.P., Roosendaal, G., Schutgens, R.E.,
Biesma, D.H., Grobbee, D.E., and Fischer, K., Clinical severity of
haemophilia A: does the classification of the 1950s still stand?
Haemophilia, 2011. 17(6): p. 849-853.
https://doi.org/10.1111/j.1365-2516.2011.02539.x PMid:21545376
- Soucie,
J.M., Monahan, P.E., Kulkarni, R., Konkle, B.A., Mazepa, M.A., and
Network, U.S.H.T.C., The frequency of joint hemorrhages and procedures
in nonsevere hemophilia A vs B. Blood Adv, 2018. 2(16): p. 2136-2144.
https://doi.org/10.1182/bloodadvances.2018020552 PMid:30143528
PMCid:PMC6113607
- Koster, T., Blann, A.D.,
Briet, E., Vandenbroucke, J.P., and Rosendaal, F.R., Role of clotting
factor VIII in effect of von Willebrand factor on occurrence of
deep-vein thrombosis. Lancet, 1995. 345(8943): p. 152-155.
https://doi.org/10.1016/S0140-6736(95)90166-3
- Rosendaal,
F.R., High levels of factor VIII and venous thrombosis. Thromb Haemost,
2000. 83(1): p. 1-2. https://doi.org/10.1055/s-0037-1613745
PMid:10669143
- Kyrle, P.A., Minar, E.,
Hirschl, M., Bialonczyk, C., Stain, M., Schneider, B., Weltermann, A.,
Speiser, W., Lechner, K., and Eichinger, S., High plasma levels of
factor VIII and the risk of recurrent venous thromboembolism. N Engl J
Med, 2000. 343(7): p. 457-462.
https://doi.org/10.1056/NEJM200008173430702 PMid:10950667
- Van
Hylckama Vlieg, A., Van Der Linden, I.K., Bertina, R.M., and Rosendaal,
F.R., High levels of factor IX increase the risk of venous thrombosis.
Blood, 2000. 95(12): p. 3678-3682.
https://doi.org/10.1182/blood.V95.12.3678 PMid:10845896
- Nossent,
A.Y., Eikenboom, J.C., and Bertina, R.M., Plasma coagulation factor
levels in venous thrombosis. Semin Hematol, 2007. 44(2): p. 77-84.
https://doi.org/10.1053/j.seminhematol.2007.01.006 PMid:17433899
- Iorio,
A., Skinner, M.W., Clearfield, E., Messner, D., Pierce, G.F., Witkop,
M., Tunis, S., and Core, H.E.M.P., Core outcome set for gene therapy in
haemophilia: Results of the coreHEM multistakeholder project.
Haemophilia, 2018. 24(4): p. e167-e172.
https://doi.org/10.1111/hae.13504 PMid:29781145
- Nogami,
K. and Shima, M., Phenotypic heterogeneity of hemostasis in severe
hemophilia. Semin Thromb Hemost, 2015. 41(8): p. 826-831.
https://doi.org/10.1055/s-0034-1395349 PMid:25615433
- Pavlova,
A. and Oldenburg, J., Defining severity of hemophilia: more than factor
levels. Semin Thromb Hemost, 2013. 39(7): p. 702-710.
https://doi.org/10.1055/s-0033-1354426 PMid:24026911
- Doshi,
B.S. and Arruda, V.R., Gene therapy for hemophilia: what does the
future hold? Ther Adv Hematol, 2018. 9(9): p. 273-293.
https://doi.org/10.1177/2040620718791933 PMid:30210756 PMCid:PMC6130099
- Nguyen,
G.N., Everett, J.K., Raymond, H., Kafle, S., Merricks, E.P., Kazazian,
H.H., Nichols, T.C., Bushman, F.D., and Sabatino, D.E., Long-Term
AAV-Mediated Factor VIII Expression in Nine Hemophilia A Dogs: A 10
Year Follow-up Analysis on Durability, Safety and Vector Integration.
2019, American Society of Hematology Washington, DC.
https://doi.org/10.1182/blood-2019-126007
- Nakai,
H., Herzog, R.W., Hagstrom, J.N., Walter, J., Kung, S.H., Yang, E.Y.,
Tai, S.J., Iwaki, Y., Kurtzman, G.J., Fisher, K.J., Colosi, P., Couto,
L.B., and High, K.A., Adeno-associated viral vector-mediated gene
transfer of human blood coagulation factor IX into mouse liver. Blood,
1998. 91(12): p. 4600-4607. https://doi.org/10.1182/blood.V91.12.4600
PMid:9616156
- Kay, M.A., Rothenberg, S.,
Landen, C.N., Bellinger, D.A., Leland, F., Toman, C., Finegold, M.,
Thompson, A.R., Read, M.S., Brinkhous, K.M., and Et Al., In vivo gene
therapy of hemophilia B: sustained partial correction in factor
IX-deficient dogs. Science, 1993. 262(5130): p. 117-119.
https://doi.org/10.1126/science.8211118 PMid:8211118
- Finn,
J.D., Ozelo, M.C., Sabatino, D.E., Franck, H.W., Merricks, E.P.,
Crudele, J.M., Zhou, S., Kazazian, H.H., Lillicrap, D., Nichols, T.C.,
and Arruda, V.R., Eradication of neutralizing antibodies to factor VIII
in canine hemophilia A after liver gene therapy. Blood, 2010. 116(26):
p. 5842-5848. https://doi.org/10.1182/blood-2010-06-288001
PMid:20876851 PMCid:PMC3031380
- Samelson-Jones,
B.J. and Arruda, V.R., Translational Potential of Immune Tolerance
Induction by AAV Liver-Directed Factor VIII Gene Therapy for Hemophilia
A. Front Immunol, 2020. 11: p. 618.
https://doi.org/10.3389/fimmu.2020.00618 PMid:32425925 PMCid:PMC7212376
- Kazazian,
H.H., Jr., An estimated frequency of endogenous insertional mutations
in humans. Nat Genet, 1999. 22(2): p. 130. https://doi.org/10.1038/9638
PMid:10369250
- Epstein, S., Bauer, S.,
Miller, A., Pilaro, A., and Noguchi, P., FDA comments on phase I
clinical trials without vector biodistribution data. Nat Genet, 1999.
22(4): p. 326. https://doi.org/10.1038/11895 PMid:10431234
- Arruda,
V.R., Fields, P.A., Milner, R., Wainwright, L., De Miguel, M.P.,
Donovan, P.J., Herzog, R.W., Nichols, T.C., Biegel, J.A., Razavi, M.,
Dake, M., Huff, D., Flake, A.W., Couto, L., Kay, M.A., and High, K.A.,
Lack of germline transmission of vector sequences following systemic
administration of recombinant AAV-2 vector in males. Mol Ther, 2001.
4(6): p. 586-592. https://doi.org/10.1006/mthe.2001.0491 PMid:11735343
- Wellman,
J., Mingozzi, F., Ozelo, M., Arruda, V., Podsakoff, G.M., Chen, Y.,
Konkle, B., Blatt, P., Hoots, K., Raffini, L., Rasko, J.E., Ragni, M.,
and High, K., Results from long-term follow-up of severe hemophilia B
subjects previously enrolled in a clinical study of AAV2-FIX gene
transfer to the liver. Molecular Therapy, 2012. 20: p. S28-S29.
https://doi.org/10.1016/S1525-0016(16)35873-7
- Boyce,
N., Trial halted after gene shows up in semen. Nature, 2001. 414(6865):
p. 677. https://doi.org/10.1038/414677a PMid:11742355
- Favaro,
P., Downey, H.D., Zhou, J.S., Wright, J.F., Hauck, B., Mingozzi, F.,
High, K.A., and Arruda, V.R., Host and vector-dependent effects on the
risk of germline transmission of AAV vectors. Mol Ther, 2009. 17(6): p.
1022-1030. https://doi.org/10.1038/mt.2009.56 PMid:19293773
PMCid:PMC2835193
- Schuettrumpf, J., Liu,
J.H., Couto, L.B., Addya, K., Leonard, D.G., Zhen, Z., Sommer, J., and
Arruda, V.R., Inadvertent germline transmission of AAV2 vector:
findings in a rabbit model correlate with those in a human clinical
trial. Mol Ther, 2006. 13(6): p. 1064-1073.
https://doi.org/10.1016/j.ymthe.2006.03.002 PMid:16631412
- Fda,
Guidance for Industry: Gene Therapy Clinical Trials - Observing
Subjects for Delayed Adverse Events, D.o.H.a.H. Services, Editor. 2006:
Rockville, MD.
- Buning, H. and Schmidt,
M., Adeno-associated Vector Toxicity-To Be or Not to Be? Mol Ther,
2015. 23(11): p. 1673-1675. https://doi.org/10.1038/mt.2015.182
PMid:26606658 PMCid:PMC4817949
- Srivastava,
A. and Carter, B.J., AAV Infection: Protection from Cancer. Hum Gene
Ther, 2017. 28(4): p. 323-327. https://doi.org/10.1089/hum.2016.147
PMid:27832705 PMCid:PMC5399746
- Donsante,
A., Miller, D.G., Li, Y., Vogler, C., Brunt, E.M., Russell, D.W., and
Sands, M.S., AAV vector integration sites in mouse hepatocellular
carcinoma. Science, 2007. 317(5837): p. 477.
https://doi.org/10.1126/science.1142658 PMid:17656716
- Donsante,
A., Vogler, C., Muzyczka, N., Crawford, J.M., Barker, J., Flotte, T.,
Campbell-Thompson, M., Daly, T., and Sands, M.S., Observed incidence of
tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther,
2001. 8(17): p. 1343-1346. https://doi.org/10.1038/sj.gt.3301541
PMid:11571571
- Gauttier, V., Pichard, V.,
Aubert, D., Kaeppel, C., Schmidt, M., Ferry, N., and Conchon, S., No
tumour-initiating risk associated with scAAV transduction in newborn
rat liver. Gene Ther, 2013. 20(7): p. 779-784.
https://doi.org/10.1038/gt.2013.7 PMid:23364314
- Bell,
P., Wang, L., Lebherz, C., Flieder, D.B., Bove, M.S., Wu, D., Gao,
G.P., Wilson, J.M., and Wivel, N.A., No evidence for tumorigenesis of
AAV vectors in a large-scale study in mice. Mol Ther, 2005. 12(2): p.
299-306. https://doi.org/10.1016/j.ymthe.2005.03.020 PMid:16043099
- Li,
H., Malani, N., Hamilton, S.R., Schlachterman, A., Bussadori, G.,
Edmonson, S.E., Shah, R., Arruda, V.R., Mingozzi, F., Wright, J.F.,
Bushman, F.D., and High, K.A., Assessing the potential for AAV vector
genotoxicity in a murine model. Blood, 2011. 117(12): p. 3311-3319.
https://doi.org/10.1182/blood-2010-08-302729 PMid:21106988
PMCid:PMC3069673
- Niemeyer, G.P., Herzog,
R.W., Mount, J., Arruda, V.R., Tillson, D.M., Hathcock, J., Van Ginkel,
F.W., High, K.A., and Lothrop, C.D., Jr., Long-term correction of
inhibitor-prone hemophilia B dogs treated with liver-directed
AAV2-mediated factor IX gene therapy. Blood, 2009. 113(4): p. 797-806.
https://doi.org/10.1182/blood-2008-10-181479 PMid:18957684
PMCid:PMC2630266
- Kaeppel, C., Beattie,
S.G., Fronza, R., Van Logtenstein, R., Salmon, F., Schmidt, S., Wolf,
S., Nowrouzi, A., Glimm, H., Von Kalle, C., Petry, H., Gaudet, D., and
Schmidt, M., A largely random AAV integration profile after LPLD gene
therapy. Nat Med, 2013. 19(7): p. 889-891.
https://doi.org/10.1038/nm.3230 PMid:23770691
- Gil-Farina,
I., Fronza, R., Kaeppel, C., Lopez-Franco, E., Ferreira, V., D'avola,
D., Benito, A., Prieto, J., Petry, H., Gonzalez-Aseguinolaza, G., and
Schmidt, M., Recombinant AAV Integration Is Not Associated With Hepatic
Genotoxicity in Nonhuman Primates and Patients. Mol Ther, 2016. 24(6):
p. 1100-1105. https://doi.org/10.1038/mt.2016.52 PMid:26948440
PMCid:PMC4923321
- Chandler, R.J., Lafave,
M.C., Varshney, G.K., Trivedi, N.S., Carrillo-Carrasco, N., Senac,
J.S., Wu, W., Hoffmann, V., Elkahloun, A.G., Burgess, S.M., and
Venditti, C.P., Vector design influences hepatic genotoxicity after
adeno-associated virus gene therapy. J Clin Invest, 2015. 125(2): p.
870-880. https://doi.org/10.1172/JCI79213 PMid:25607839 PMCid:PMC4319425
- Nault,
J.C., Datta, S., Imbeaud, S., Franconi, A., Mallet, M., Couchy, G.,
Letouze, E., Pilati, C., Verret, B., Blanc, J.F., Balabaud, C.,
Calderaro, J., Laurent, A., Letexier, M., Bioulac-Sage, P., Calvo, F.,
and Zucman-Rossi, J., Recurrent AAV2-related insertional mutagenesis in
human hepatocellular carcinomas. Nat Genet, 2015. 47(10): p. 1187-1193.
https://doi.org/10.1038/ng.3389 PMid:26301494
- Berns,
K.I., Byrne, B.J., Flotte, T.R., Gao, G., Hauswirth, W.W., Herzog,
R.W., Muzyczka, N., Vandendriessche, T., Xiao, X., Zolotukhin, S., and
Srivastava, A., Adeno-Associated Virus Type 2 and Hepatocellular
Carcinoma? Hum Gene Ther, 2015. 26(12): p. 779-781.
https://doi.org/10.1089/hum.2015.29014.kib PMid:26690810
PMCid:PMC4809064
- Berns, K.I. and
Muzyczka, N., AAV: An Overview of Unanswered Questions. Hum Gene Ther,
2017. 28(4): p. 308-313. https://doi.org/10.1089/hum.2017.048
PMid:28335618 PMCid:PMC5399733
- Logan,
G.J., Dane, A.P., Hallwirth, C.V., Smyth, C.M., Wilkie, E.E., Amaya,
A.K., Zhu, E., Khandekar, N., Ginn, S.L., Liao, S.H.Y., Cunningham,
S.C., Sasaki, N., Cabanes-Creus, M., Tam, P.P.L., Russell, D.W.,
Lisowski, L., and Alexander, I.E., Identification of liver-specific
enhancer-promoter activity in the 3' untranslated region of the
wildtype AAV2 genome. Nat Genet, 2017. 49(8): p. 1267-1273.
https://doi.org/10.1038/ng.3893 PMid:28628105
- George,
L.A., Ragni, M.V., Samelson-Jones, B.J., Cuker, A., Runoski, A.R.,
Cole, G., Wright, F., Chen, Y., Hui, D.J., Wachtel, K., Takefman, D.,
Couto, L.B., Reape, K.Z., Carr, M.E., Anguela, X.M., and Katherine A.
High, M., Spk-8011: Preliminary Results from a Phase 1/2 Dose
Escalation Trial of an Investigational AAV-Mediated Gene Therapy for
Hemophilia A. Blood, 2017. 130(Suppl1): p. 604.
- George,
L.A., Ragni, M.V., Rasko, J.E.J., Raffini, L.J., Samelson-Jones, B.J.,
Ozelo, M., Hazbon, M., Runowski, A.R., Wellman, J.A., Wachtel, K.,
Chen, Y., Anguela, X.M., Kuranda, K., Mingozzi, F., and High, K.A.,
Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV
for Gene Transfer: AAV2-hFIX16 for Severe Hemophilia B. Mol Ther, 2020.
https://doi.org/10.1016/j.ymthe.2020.06.001
- Ema, Guideline on follow-up of patients administered with gene therapy medicinal products. 2009: London, UK. p. 1-12.
- Mount,
J.D., Herzog, R.W., Tillson, D.M., Goodman, S.A., Robinson, N.,
Mccleland, M.L., Bellinger, D., Nichols, T.C., Arruda, V.R., Lothrop,
C.D., Jr., and High, K.A., Sustained phenotypic correction of
hemophilia B dogs with a factor IX null mutation by liver-directed gene
therapy. Blood, 2002. 99(8): p. 2670-2676.
https://doi.org/10.1182/blood.V99.8.2670 PMid:11929752
- Van
Dijk, K., Van Der Bom, J.G., Bax, K.N., Van Der Zee, D.C., and Van Den
Berg, M.H., Use of implantable venous access devices in children with
severe hemophilia: benefits and burden. Haematologica, 2004. 89(2): p.
189-194.
- Hay, C.R., Dimichele, D.M., and
International Immune Tolerance, S., The principal results of the
International Immune Tolerance Study: a randomized dose comparison.
Blood, 2012. 119(6): p. 1335-1344.
https://doi.org/10.1182/blood-2011-08-369132 PMid:22101900
- Arruda,
V.R., Doshi, B.S., and Samelson-Jones, B.J., Novel approaches to
hemophilia therapy: successes and challenges. Blood, 2017. 130(21): p.
2251-2256. https://doi.org/10.1182/blood-2017-08-742312 PMid:29018078
PMCid:PMC5813735
- Stanford, S., Pink, R.,
Creagh, D., Clark, A., Lowe, G., Curry, N., Pasi, J., Perry, D., Fong,
S., Hayes, G., Chandrakumaran, K., and Rangarajan, S.,
Adenovirus-associated antibodies in UK cohort of hemophilia patients: A
seroprevalence study of the presence of adenovirus-associated virus
vector-serotypes AAV5 and AAV8 neutralizing activity and antibodies in
patients with hemophilia A. Res Pract Thromb Haemost, 2019. 3(2): p.
261-267. https://doi.org/10.1002/rth2.12177 PMid:31011710
PMCid:PMC6462753
- Aronson, S.J., Veron,
P., Collaud, F., Hubert, A., Delahais, V., Honnet, G., De Knegt, R.J.,
Junge, N., Baumann, U., Di Giorgio, A., D'antiga, L., Ginocchio, V.M.,
Brunetti-Pierri, N., Labrune, P., Beuers, U., Bosma, P.J., and
Mingozzi, F., Prevalence and Relevance of Pre-Existing
Anti-Adeno-Associated Virus Immunity in the Context of Gene Therapy for
Crigler-Najjar Syndrome. Hum Gene Ther, 2019. 30(10): p. 1297-1305.
https://doi.org/10.1089/hum.2019.143 PMid:31502485 PMCid:PMC6763963
- Ertl,
H.C.J. and High, K.A., Impact of AAV Capsid-Specific T-Cell Responses
on Design and Outcome of Clinical Gene Transfer Trials with Recombinant
Adeno-Associated Viral Vectors: An Evolving Controversy. Hum Gene Ther,
2017. 28(4): p. 328-337. https://doi.org/10.1089/hum.2016.172
PMid:28042943
- Unzu, C., Hervas-Stubbs,
S., Sampedro, A., Mauleon, I., Mancheno, U., Alfaro, C., De Salamanca,
R.E., Benito, A., Beattie, S.G., Petry, H., Prieto, J., Melero, I., and
Fontanellas, A., Transient and intensive pharmacological
immunosuppression fails to improve AAV-based liver gene transfer in
non-human primates. J Transl Med, 2012. 10: p. 122.
https://doi.org/10.1186/1479-5876-10-122 PMid:22704060 PMCid:PMC3412719
- Monteilhet,
V., Saheb, S., Boutin, S., Leborgne, C., Veron, P., Montus, M.F.,
Moullier, P., Benveniste, O., and Masurier, C., A 10 patient case
report on the impact of plasmapheresis upon neutralizing factors
against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther,
2011. 19(11): p. 2084-2091. https://doi.org/10.1038/mt.2011.108
PMid:21629225 PMCid:PMC3222518
- Mimuro,
J., Mizukami, H., Hishikawa, S., Ikemoto, T., Ishiwata, A., Sakata, A.,
Ohmori, T., Madoiwa, S., Ono, F., Ozawa, K., and Sakata, Y., Minimizing
the inhibitory effect of neutralizing antibody for efficient gene
expression in the liver with adeno-associated virus 8 vectors. Mol
Ther, 2013. 21(2): p. 318-323. https://doi.org/10.1038/mt.2012.258
PMid:23247100 PMCid:PMC3594013
- Leborgne,
C., Barbon, E., Alexander, J.M., Hanby, H., Delignat, S., Cohen, D.M.,
Collaud, F., Muraleetharan, S., Lupo, D., Silverberg, J., Huang, K.,
Van Wittengerghe, L., Marolleau, B., Miranda, A., Fabiano, A.,
Daventure, V., Beck, H., Anguela, X.M., Ronzitti, G., Armour, S.M.,
Lacroix-Desmazes, S., and Mingozzi, F., IgG-cleaving endopeptidase
enables in vivo gene therapy in the presence of anti-AAV neutralizing
antibodies. Nat Med, 2020. 26(7): p. 1096-1101.
https://doi.org/10.1038/s41591-020-0911-7 PMid:32483358
- Elmore,
Z.C., Oh, D.K., Simon, K.E., Fanous, M.M., and Asokan, A., Rescuing AAV
gene transfer from antibody neutralization with an IgG-degrading
enzyme. bioRxiv, 2020: p. 2020.2005.2012.092122.
https://doi.org/10.1101/2020.05.12.092122
- Jordan,
S.C., Lorant, T., and Choi, J., IgG Endopeptidase in Highly Sensitized
Patients Undergoing Transplantation. N Engl J Med, 2017. 377(17): p.
1693-1694. https://doi.org/10.1056/NEJMoa1612567
- Meliani,
A., Boisgerault, F., Hardet, R., Marmier, S., Collaud, F., Ronzitti,
G., Leborgne, C., Costa Verdera, H., Simon Sola, M., Charles, S.,
Vignaud, A., Van Wittenberghe, L., Manni, G., Christophe, O.,
Fallarino, F., Roy, C., Michaud, A., Ilyinskii, P., Kishimoto, T.K.,
and Mingozzi, F., Antigen-selective modulation of AAV immunogenicity
with tolerogenic rapamycin nanoparticles enables successful vector
re-administration. Nat Commun, 2018. 9(1): p. 4098.
https://doi.org/10.1038/s41467-018-06621-3 PMid:30291246
PMCid:PMC6173722
- Machin, N., Ragni, M.V.,
and Smith, K.J., Gene therapy in hemophilia A: a cost-effectiveness
analysis. Blood Adv, 2018. 2(14): p. 1792-1798.
https://doi.org/10.1182/bloodadvances.2018021345 PMid:30042145
PMCid:PMC6058236
- Lang, J.F., Toulmin,
S.A., Brida, K.L., Eisenlohr, L.C., and Davidson, B.L., Standard
screening methods underreport AAV-mediated transduction and gene
editing. Nat Commun, 2019. 10(1): p. 3415.
https://doi.org/10.1038/s41467-019-11321-7 PMid:31363095
PMCid:PMC6667494
- Skinner, M.W., Nugent,
D., Wilton, P., O'mahony, B., Dolan, G., O'hara, J., and Berntorp, E.,
Achieving the unimaginable: Health equity in haemophilia. Haemophilia,
2020. 26(1): p. 17-24. https://doi.org/10.1111/hae.13862 PMid:31724316
[TOP]