Vincenzo de Sanctis1, Ashraf T Soliman2, Shahina Daar3, Ploutarchos Tzoulis4, Salvatore Di Maio5 and Christos Kattamis6.
1 Coordinator
of ICET-A Network (International Network of Clinicians for
Endocrinopathies in Thalassemia and Adolescence Medicine), Ferrara,
Italy.
2 Department of Pediatrics, Division of
Endocrinology, Hamad General Hospital, Doha, Qatar and Department of
Pediatrics, Division of Endocrinology, Alexandria University Children’s
Hospital, Alexandria, Egypt.
3 Department of Haematology, College of Medicine and Health Sciences, Sultan Qaboos University, Sultanate of Oman.
4 Department of Diabetes & Endocrinology, Whittington Hospital, University College London, London, UK.
5 Emeritus Director in Pediatrics, Children’s Hospital “Santobono-Pausilipon,” Naples, Italy.
6 First Department of Paediatrics, National Kapodistrian University of Athens, Greece.
Correspondence to: Vincenzo de Sanctis. Coordinator of ICET-A
Network (International Network of Clinicians for Endocrinopathies in
Thalassemia and Adolescence Medicine), Ferrara, Italy. E-mail:
vdesanctis@libero.it
Published: March 1, 2021
Received: November 15, 2020
Accepted: February 11, 2021
Mediterr J Hematol Infect Dis 2021, 13(1): e2021021 DOI
10.4084/MJHID.2021.021
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Objective:
To study the endocrine pancreas' function in transfusion-dependent
β-thalassemia (β-TDT) patients with a normal glucose tolerance test
(NGT) and hypoinsulinemia. In addition, the prospective long–term
follow-up using an annual oral glucose tolerance test (OGTT) to detect
any abnormality of glucose metabolism.
Patients and methods:
Seven β-TDT patients (mean age 22.4 ± 4.2 years) with NGT and
inadequate insulin response (hypoinsulinemia) to OGTT were referred for
a second opinion to an Italian Centre.
Results:
The first-phase insulin response (FPIR), expressed as the sum of 1 and
3 minutes insulin, to intravenous glucose tolerance test (IVGTT), was
between the 1st and 3rd percentile in two patients and between the 3rd
and 10th percentile in five. The results were not associated with
β-cell autoimmunity. After 43 ± 26 months (range 11 - 80 months) of
follow-up, two patients developed impaired glucose tolerance (IGT),
three both IGT and impaired fasting glucose (IFG) and two overt
diabetes mellitus (DM). Interestingly, the patients who developed DM
had, at baseline, the lowest value of the insulinogenic index (IGI:
0.08 and 0.25), defined as the ratio of the increment of plasma insulin
to plasma glucose during the first 30 minutes after OGTT. Moreover, a
significant correlation was found between the IGI at baseline and at
follow-up in the patients who developed IGT with or without IFG (R=
0.927; P: 0.023). A significant reduction of Matsuda insulin
sensitivity index (ISIM) and Insulin Secretion-Sensitivity Index-2
(ISSI-2) was documented in the study cohort at the diagnosis of IFG,
IGT, and DM. There was a significant inverse correlation between ISSI-2
and area under the curve plasma glucose (AUC-PG).
Conclusions:
These data demonstrated, for the first time, progressive deterioration
in glucose homeostasis in β-TDT subjects with NGT and hypoinsulinemia
and that the ISSI-2 index may be a valuable parameter to identify
patients at high risk for developing glucose dysregulation.
|
Introduction
β-thalassemias
are amongst the commonest genetic disorders worldwide, caused by a
reduction of the β-globin chains of the hemoglobin molecule, leading to
severe chronic hemolytic anemia. Transfusion-dependent β-thalassemia
(β-TDT/β-thalassemia major) patients present to pediatric departments
in infancy and early childhood (< 3 years) with severe anemia that
requires lifelong regular transfusions for survival. The disease
process's culprit is the secondary iron overload from regular
transfusions, which may lead to organ damage and failure, mainly
involving the heart, liver, and endocrine glands. Excess iron is
removed from the body by using parenteral or oral iron chelators
starting from early childhood; using iron chelators regularly is
difficult for many caregivers and patients, leading to poor compliance.[1]
In patients receiving suboptimal iron chelation, pancreatic iron loading starts in early childhood.[2]
The pathogenesis of glycemic abnormalities in β-TDT patients is complex
and multifactorial, and significantly different from the pathogenesis
of glucose metabolism's dysregulation in a normal individual,
particularly children and adolescents. Several studies have shown that
insulin resistance and insulin deficiency mark both the prediabetic
state and diabetes in thalassemia.[3-5] Insulin secretory defects, however, may originate from pancreatic β-cell damage rather than from insulin resistance.[6]
To evaluate the possible role of autoimmunity in the pathogenesis of diabetes, Monge et al.[7]
studied a cohort of 53 β-TDT patients, including twelve patients with
diabetes (22.6%). To be evaluated about the activation of an autoimmune
response, individuals were tested for islet cell antibodies (ICA),
glutamic acid decarboxylase (GAD) autoantibodies, insulin
autoantibodies (IAA), and serum antinuclear antibodies (ANA). The study
demonstrated evidence of immune system activation against pancreatic
β-cells in β-TDT patients. The authors suggested that iron deposition
may act through oxidative damage as an environmental factor that
triggers the autoimmune response.[7]
Overall,
these diabetogenic factors have a cumulative effect that seems to be
progressive and can lead to glucose intolerance in long-term.[8]
In
the light of these observations, we report seven β-TDT patients with
normal glucose tolerance test (NGT) and reduced insulin response
(hypoinsulinemia), after an oral glucose tolerance test (OGTT),
referred for a second opinion, from Italian Centers taking care of
patients with hemoglobinopathies. Due to very uncommon observations,
their long-term natural history, assessed by an annual OGTT, is
reported with the aim of detecting any abnormality of glucose
metabolism.
Patients and Methods
At baseline.
Our cross-sectional study started in 2008. Seven Italian Centers,
taking care of patients with hemoglobinopathies, referred for a second
opinion to an ICET-A (International Network of Clinicians for
Endocrinopathies in Thalassemia and Adolescence Medicine) Italian
Center, seven β-TDT patients (mean age 22.4 ± 4.2 years) with NGT
associated to hypoinsulinemia during OGTT. The long-term patients'
natural history of OGTT is also reported. According to Crofts et al.,[9]
the insulin secretory capacity was defined as reduced if all insulin
values during OGTT were ≤ 30 μU/mL. Based upon plasma glucose results
on OGTT, patients were classified according to the American Diabetes
Association (ADA) criteria.[10]
The following
data were collected from each subject: demographic data, age at first
transfusion, the interval between transfusions, type and compliance to
iron chelation therapy, anthropometric data [weight, height, body mass
index (BMI)], pubertal status, and associated endocrine complications.
BMI was calculated as body weight in kilograms divided by height in
meters squared.
A subject was considered obese when BMI exceeded 30 Kg/m2,
overweight when BMI was 25 - 30 kg/m², of average weight when BMI was
18.5-25 kg/m², and underweight when the BMI was < 18.5 kg/m².
First step:
Study of autoimmunity and first-phase insulin release (FPIR) after
intravenous glucose tolerance test (IVGTT). The β-cell autoimmunity was
assessed in all patients by GAD, ICA, and IAA. The samples were
analyzed for ICA by immunofluorescence and for GAD and IAA by specific
radioligand binding assays. Waiting for autoimmunity results against
pancreatic β-cells, an intravenous glucose tolerance test (IVGTT) was
performed between 08.30 and 09.30, after fasting for 8-10 hours.
Patients consumed a regular diet for 3 days and avoided smoking before
the test.
An intravenous catheter was placed in the antecubital
vein, and an intravenous bolus of 0.5 g glucose/kg body weight (maximum
35 g, as 25% water solution) was injected manually through an
indwelling intravenous cannula in the contralateral antecubital vein
over 3 - 4 minutes. The end of glucose infusion was defined as time
zero. Blood was assessed for insulin radioimmunoassay at baseline
(completion of glucose infusion) and after 1, 3, 5, and 10 minutes. The
sum of insulin values at 1 and 3 minutes was used as an index of the
first phase insulin response (FPIR).[11,12] For FPIR, the 1st percentile is 48 µU/ml, 3rd percentile 56 µU/ml, 5th percentile 64 µU/ml, 10th percentile 81 µU/ml, and 50th percentile 162 µU/ml.[12]
Other laboratory assays and cardiac imaging.
The serum alanine aminotransferase (ALT) level was determined by an
automated analyzer (normal range 0–40 mU/L). Serum ferritin was
measured by immunoassays. The 90th percentile of reported average values is 201-243: ng/mL.[13]
Plasma
glucose was measured using an automated glucose oxidase reaction
(Glucose Analyser, Ames). Plasma samples were centrifuged at 4°C,
separated, and stored at −20°C
until assay. Plasma insulin was determined by a commercial solid-phase
radioimmunoassay technique (Coat-A-Count insulin kit, Diagnostic
Products Corporation, Los Angeles, CA) with intra- and inter-assay
coefficients of variance of 3.3% and 2.5%, respectively.
Cardiac
T2* was assessed by magnetic resonance imaging (MRI) using a 1.5 T
scanner. A conservative cut-off value of heart T2* > 20 ms was
considered normal.[14]
Follow-up.
After IVGTTs, all patients were followed yearly with an OGTT to detect
any potential glucose abnormalities, including quantitative and
qualitative defects in insulin secretion, using the following methods
and calculation indices;
Plasma glucose, insulin, and islet β-cell function indices from the OGTT.
One week following the blood transfusion, a 75-g OGTT was performed in
the morning after an overnight fast. Blood samples were collected from
the venous catheter at 0, 30, 60, 90, 120, and 180 minutes for plasma
glucose and insulin assessment. Participants remained seated for the
entire testing period.
Based upon plasma glucose results on
OGTT, patients were classified according to the American Diabetes
Association (ADA) criteria[10] into the following categories:
- Normoglycemia: FPG < 100 mg/dL and 2-h PG < 140 mg/dL;
- IFG: FPG between 100 and 125 mg/dL;
- IGT: 2-h PG between 140 and 199 mg/dL;
- DM: Fasting plasma glucose (FPG) ≥ 126 mg/dl or 2-hour plasma glucose (2-h PG) ≥ 200 mg/dL.
Different
indirect indices were applied for insulin resistance and sensitivity
recognition; among them those calculated from fasting glucose and
insulin concentration and those derived during the OGTT, including
Insulinogenic Index (IGI), plasma glucose (PG), and insulin (INS) area
under the curves (AUC- PG 0–120 min and AUC-INS 0–120 min), Homeostasis
Model Assessment of Insulin Resistance (HOMA1-IR), Quantitative Insulin
sensitivity Check Index (QUICKI), Matsuda insulin sensitivity index
(ISIM), and Insulin Secretion-Sensitivity Index-2 (ISSI-2). AUC- PG,
and AUC-INS during OGTT were calculated with the trapezoid method.[15-19]
Statistical analysis.
Data are presented as means ± standard deviation (SD). Statistical
comparison between parameters was made using the paired “t” test.
Simple linear regression tested the correlations between variables. For
the statistical analysis, a software program was used and validated,
according to Alder and Roesser.[20] A p-value < 0.05 was considered statistically significant.
Ethics.
All procedures were in accordance with the 1964 Helsinki declaration
and its later amendments. According to the Italian regulations, the
local Ethics Committee's approval was not required for the following
reasons: no identifiable private information was collected; patients
underwent only routine diagnostic and therapeutic procedures according
to current guidelines;[10] an anonymized dataset was
analyzed. Informed consent was obtained from all patients after a
detailed explanation of the study's nature and purpose and the likely
risks and benefits of study participation.
Results
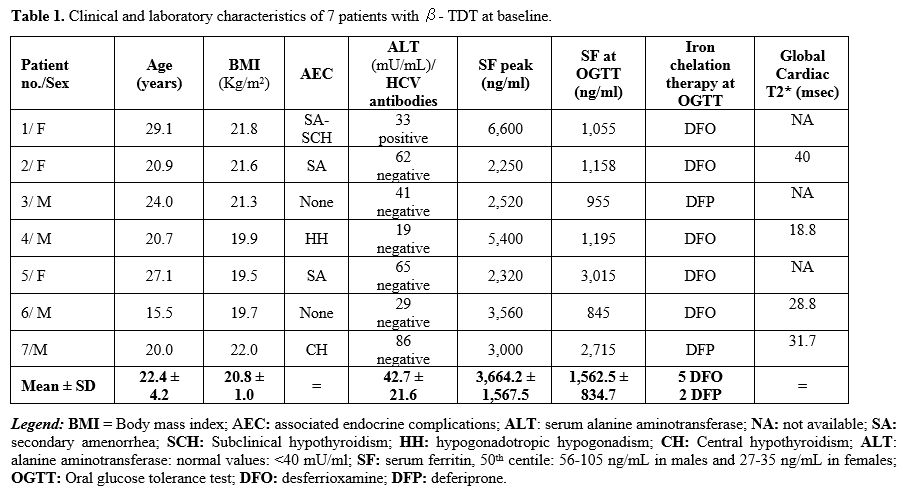
Clinical and laboratory characteristics. The patients' demographic data and other parameters are shown in Table 1.
All patients were on regular blood transfusions and iron chelation
therapy. They were neither overweight/obese nor underweight.
 |
Table
1. Clinical and laboratory characteristics of 7 patients with β- TDT at baseline.
|
Five
of seven patients were on desferrioxamine (DFO) monotherapy with an
average daily dose of 40 ± 5.34 mg/kg body weight (range: 35-50 mg/kg
body weight), given subcutaneously by pump for 7 to 8 hours per night,
for 5 to 6 days a week. Two patients were on oral deferiprone (DFO)
monotherapy (total dose 75 mg/kg/day, divided into 3 doses). At
baseline, their mean serum ferritin levels were 1,562.5 ± 834.7 ng/mL
(range: 955-3,015 ng/mL) (Table 1).
The
highest mean serum ferritin level ("peak level") registered in the
course of previous years was 3,664.2 ± 1,567.5 ng/mL (range:
6,000-2,250 ng/mL). ALT was elevated in 3 patients (patients 2, 5, and
7 with DFO). Cardiac T2* was impaired in one patient (18.8 msec) with a
peak ferritin level of 5,400 ng/mL.
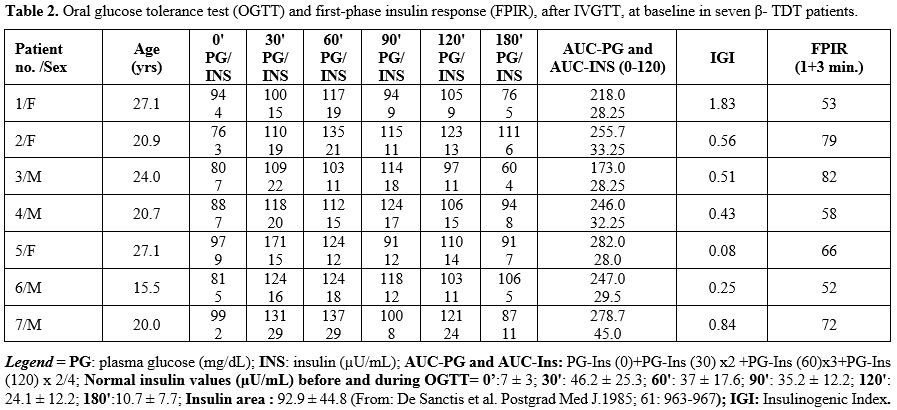
Glucose tolerance at baseline.
At baseline, all patients had normal glucose values with reduced
insulin secretion on OGTT. The peak of plasma glucose and insulin
response, after OGTT, was delayed (60'-90' minutes) in 3 patients (Table 2).
 |
Table
2. Oral glucose tolerance test (OGTT) and first-phase insulin response
(FPIR), after IVGTT, at baseline in seven β- TDT patients.
|
No
statistical correlation was observed between age and BMI, AUC-PG,
AUC-Ins, and peak of serum ferritin level. Similarly, a non-significant
correlation was found between HOMA-IR, QUICKI, and ISSI-2 with AUC-PG
and AUC-INS. Conversely, a significant correlation was found between
ISIM and AUC-INS (R= 0.7824; P: 0.037) but not with AUC-PG. An inverse
correlation was present between HOMA-IR and QUICKI (R= -0.9567; P:
0.0007).
Correlations between serum ferritin levels (peak over
previous years ) with HOMA-IR, QUIKI, AUC-PG, and AUC-INS were not
significant. An inverse correlation was present between serum ferritin
at the time of study vs. ISIM and ISSI-2 (R= -0.962; P: 0.00053 and R=
-0.8459; P: 0.016, respectively).
Adverse effects of OGTT. There were no adverse events secondary to OGTT, and none of the subjects experienced hypoglycemia.
FPIR after IVGTT.
In two patients, the FPIR value FPIR after IVGTT. In two patients, the
FPIR value (expressed as the sum of 1 + 3 min insulin) was between the 1st and 3rd percentile (patients 1 and 6), and in five patients between the 3rd and 10th percentile (Table 2).
No correlation was observed between FPIR and age, AUC-PG, AUC-Ins, the
peak of serum ferritin level, or serum ferritin at baseline. These
results were not associated with β-cell autoimmunity.
Glucose tolerance, insulin and islet β-cell function indices from the OGTT during annual follow-up.
During the annual OGTT follow-up, glucose homeostasis's first
alteration was documented after 43 ± 26 months (range 13-80). Two
patients developed IGT (patients 4 and 7), three IFG associated with
IGT (patients 1, 2 and 3), and two overt DM (patients 5 and 6) within
3.3 and 1.1 yrs, respectively (Table 3). All seven patients had abnormal 2-hour plasma glucose (Figure 1).
A strong linear correlation was also observed between the FPIR value at
baseline and interval (expressed in months) of the appearance of
glucose homeostasis abnormalities (R= 0.8753, P: 0.009).
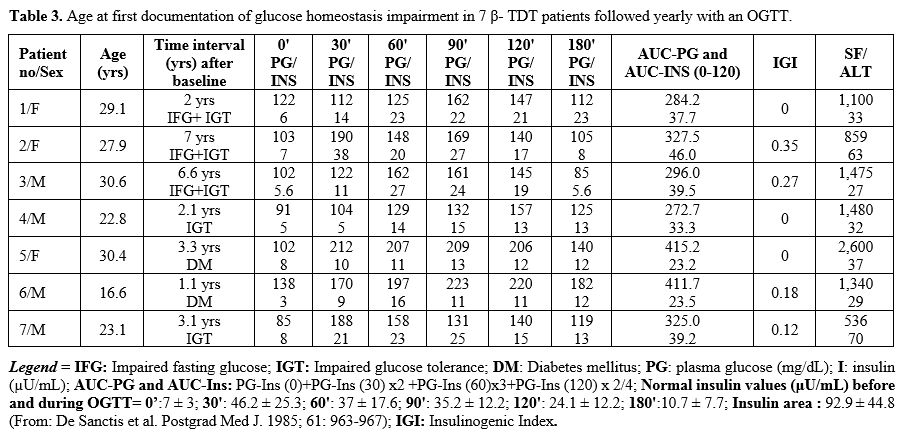 |
Table 3. Age at first documentation of glucose homeostasis impairment in 7 β- TDT patients followed yearly with an OGTT. |
 |
Figure 1. Outcome of 7 β-TDT patients with normal OGTT and hypoinsulinemia followed for 13-80 months. |
The
AUC-Ins120/AUC-PG 120 was not statistically different between baseline
values vs. last observation (P: 0.16). However, the β-TDT patients who
developed DM had the lowest IGI index values at baseline (0.08 and
0.25). Throughout the entire follow-up, five patients developed IGT;
there was a significant difference in baseline IGI in these patients
compared to those who did not develop IGT (R= 0.927; P: 0.023),
independently from IFG presence.
There was no significant change
in HOMA-IR and QUICKI during the follow-up. On the other hand, a
significant reduction in the ISIM and ISSI-2 was documented in the
whole study cohort at IFG-IGT and DM diagnosis. There was a significant
inverse correlation between ISSI-2 and AUC-PG (R= -0.9617, P: 0.0005) (Table 4).
 |
Table 4. Follow-up of
indirect indices of insulin secretion and resistance at baseline and at
the diagnosis of IGT and DM in patients with β- TDT.
|
No
significant BMI changes occurred in patients from the baseline to the
end of the follow-up period (20.8 ±1.0 vs. 21.1 ± 1.2 Kg/m2;
P: NS). The mean serum ferritin level did not differ at baseline or at
the occurrence of IGT/IFG/DM (1,562 ± 901.6 vs. 1,341 ± 653.3 ng/mL; P:
NS).
Due to the limited number of β-TDT patients, a comparison of collected parameters between males and females was impossible.
Discussion
Disturbances
of glucose tolerance occur in a significant number of adolescent and
adult patients with β-TDT who are at high risk for developing abnormal
glucose handlings, such as IFG, IGT, and DM. These glucose disturbances
are mainly due to decreased insulin secretion and insulin resistance
(IR) secondary to iron overload.[1-6]
OGTT is widely used in clinical settings to diagnose patients with IGT and DM, based on ADA recommendations.[10]
OGTT is a valuable tool in diabetes research and is commonly used for
screening, evaluating disease progression, monitoring treatment, and
thoroughly studying physiological and patho-physiological conditions.[21]
Because of the increased risk of glucose disturbances in β-TDT patients
with iron overload, there are recommendations for annual evaluation of
glycemic status in children over ten years.[22]
However,
OGTT gives only a crude estimate of β-cell secretory function. The
initial insult is considered insulin resistance, which, as long as it
is compensated by hyperinsulinemia, does not lead to hyperglycemia.
Finally, when pancreatic β-cells are unable to secrete an increased
amount of insulin, to compensate for insulin resistance, hyperglycemia
develops.[3-6]
In this study, patients with
β-TDT, despite exhibiting normal glucose tolerance as defined by
standard glucose homeostasis criteria, presented with impaired insulin
secretion (hypoinsulinemia) during OGTT. The reduced insulin response
to OGTT was further confirmed by FPIR values (between the 1st and 3rd percentile in two patients and in five between the 3rd and 10th
percentile). The potential role of autoimmunity was excluded by the
assessment of islet cell antibodies (ICA), glutamic acid decarboxylase
(GAD) autoantibodies, and insulin autoantibodies (IAA).
Although
we cannot identify the cause of the low insulin levels found during
OGTT at baseline, we can speculate that these patients had a higher
insulin sensitivity. However, we cannot exclude, on the basis of
long-term follow-up, the decreased β-cell secretory capacity was due to
an initial and progressive impairment of pancreatic β-cells toxicity of
iron overload,[6] and/or to loss of pancreatic micro-vascularity.[23-25]
A
strong linear correlation was observed between the FPIR value at
baseline and the time interval of the appearance of glucose
abnormalities (R= 0.8753, P: 0.009). In two patients, the FPIR value
(expressed as the sum of 1 + 3 min insulin) was between the 1st and 3rd percentile (patients 1 and 6), and in five, between the 3rd and 10th
centile. Nevertheless, the insulin response to IVGTT in all patients
was higher compared to OGTT. In healthy individuals, the incretin
effect accounts for 70% of the insulin response after oral glucose
administration. However, we can speculate a possible reduction of the
incretin effect in pivotal experiments; to assess the incretin effects
the comparison between oral and i.v. glucose stimuli were performed
using well-matched glucose concentrations. That was not the case in our
subjects.
From the baseline, all patients were followed yearly
with an OGTT. After a total period of 43 ± 26 months (range:11-80) of
follow-up, two patients developed IGT, three both IFG and IGT, and two
developed DM. Nevertheless, no statistical difference was found between
the AUC-Ins120/ AUC-PG120, considered an index of total insulin
secretion during the OGTT. This observation suggests that a decrease in
insulin sensitivity would be a factor involved in the deterioration of
glucose tolerance when the insulin secretion is compromised. In
patients with β-TDT, IR may be due to: (a) a direct effect of iron
overload to pancreatic β-cells and/or (b) hepatic dysfunction leading
to reduced hepatic clearance of insulin resulting in impaired glucose
homeostasis.[26-27] Moreover, IR can be related to the variation of qualitative and quantitative nutritional intake and low physical activity.
Several
techniques have been used in humans to assess IR. Methods for
quantifying β-cell sensitivity to glucose (hyperglycemic clamp
technique) and tissue sensitivity to insulin (euglycemic insulin clamp
technique) are generally recognized as the gold standard reference for
assessing IR. However, these methods are laborious, expensive, and
inconvenient to patients or study subjects and are not routinely
available to every physician. Since OGTT is cheap and straightforward,
many mathematical models’ formulas have been developed using OGTT
parameters. We calculated the most widely used surrogate indices: IGI,
HOMA-IR, QUICKI, ISIM, and ISSI-2, calculated from fasting glucose and
insulin concentration, and those derived OGTT evaluation.[28-30]
In
brief, IGI is calculated as the ratio of plasma insulin's increment to
glucose concentration 30 min after an OGTT. The loss of this early
insulin release is a feature of the prediabetic condition. HOMA and
QUICKI are mathematically related and provide essentially identical
information. Both primarily reflect hepatic insulin resistance rather
than peripheral insulin resistance.[31]
ISIM
combines two terms that account for insulin sensitivity of the hepatic
as well as the peripheral tissues. One part of the equation consists of
a hyperbolic conversion of the product of fasting plasma glucose and
insulin as a measure of hepatic sensitivity. The second accounts for
whole-body insulin sensitivity, described by the inverse product of the
mean glucose and insulin concentration after the glucose load. ISSI-2
is defined as the ratio of the area under the insulin curve to the area
under the glucose curve, multiplied by the Matsuda index; it
constitutes a surrogate measure of insulin secretion relative to
insulin sensitivity and emphasizes the pivotal role of impaired insulin
secretion in the development of dysregulation of glucose homeostasis.
Substantially, it refers to the relationship between insulin
sensitivity and insulin secretion.[15-19]
Our
results confirm that the ISSI-2 index may be valuable parameters to
identify β -TDT patients at the highest risk for developing glucose
dysregulation.[32]
Interestingly, the β -TDT
patients who developed DM had, at baseline, the lowest values of the
IGI index (0.08 and 0.25). Moreover, a significant correlation was
found between IGI at baseline in those patients who developed IGT
compared to those who did not, regardless of IFG (R= 0.927; P:
0.023416). No significant changes in HOMA-IR and QUICKI were observed
during follow-up. Conversely, a significant reduction of ISIM and
ISSI-2 was documented in the whole study cohort at the diagnosis of
IFG-IGT and DM, with a significant inverse correlation between ISSI-2
and AUC-PG. Therefore, ISIM and ISSI-2, which include post-load glucose
and insulin concentrations, provided a more accurate estimate of
whole-body insulin sensitivity than HOMA-IR or QUICKI, derived from
fasting measurements only, thus constituting a more sensitive tool for
detecting alterations of glucose sensitivity/resistance in β-TDT
patients.
Our study has several limitations: a) the small number
of patients enrolled in the study, but this seems inevitable as the
profile of glucose dysregulation in TDT is reported for the first time
and is very rare; b) the use of surrogate indices for assessing insulin
sensitivity, and c) the evaluation of iron overload assessed with serum
ferritin not associated to the evaluation of pancreatic iron stores by
magnetic resonance imaging (MRI). However, the major strength is the
long-term follow-up study of an uncommon group of β-TDT patients with
NGT and hypoinsulinemia who developed, after 43 ± 26 months, a glucose
dysregulation.
Conclusions
There
are limited data, if any, about the function of the endocrine pancreas
in normoglycemic β-TDT patients with impaired insulin secretion on
OGTT. In our experience, IVGTT, a test that nowadays is not routinely
included in the screening of glucose metabolism disturbances in β-TDT
patients, could be useful in selected patients with NGT and reduced
insulin response (hypoinsulinemia) after OGTT. The longitudinal
evaluation of surrogate measures of insulin secretion and/or
sensitivity allowed us to demonstrate that a weakening of peripheral
insulin action may contribute to the development of glucose homeostasis
impairment. However, a larger number of patients is necessary to
understand better the respective roles of the progressive reduction of
insulin secretion and the variation of insulin sensitivity. Our
observations also support the need for a continuous follow-up with
regular OGTT and timed counseling to promote lifestyle changes in
high-risk subjects. As demonstrated in the general population and
patients with type 2 diabetes, physical activity programs, diet
changes, and pharmacological interventions could be useful measures to
improve glucose tolerance in patients with β-TDT. Further studies are
required to determine the possible benefits of insulin oral
secretagogues and to establish the best treatment for this patient
group.
References
- De Sanctis V, Soliman AT, Elsedfy H, Pepe A,
Kattamis C, El Kholy M, Yassin M. Diabetes and Glucose Metabolism in
Thalassemia Major: An Update. Expert Rev Hematol. 2016;9:401-408. https://doi.org/10.1586/17474086.2016.1136209 PMid:26697756
- Liang
Y, Bajoria R, Jiang Y, Su H, Pan H, Xia N, Chatterjee R, Lai Y.
Prevalence of diabetes mellitus in Chinese children with thalassaemia
major. Trop Med Int Health. 2017;22:716-724. https://doi.org/10.1111/tmi.12876 PMid:28544032
- Dmochowski
K, Finegood DT, Francombe W, Tyler B, Zinman B. Factors determining
glucose tolerance in patients with thalassemia major. J Clin Endocrinol
Metab. 1993;77:478-483. https://doi.org/10.1210/jcem.77.2.8345055 PMid:8345055
- Merkel
PA, Simonson DC, Amiel SA, Plewe G, Sherwin RS, Pearson HA, Tamborlane
WV. Insulin resistance and hyperinsulinemia in patients with
thalassemia major treated by hypertransfusion. N Engl J Med.
1988;318:809-814. https://doi.org/10.1056/NEJM198803313181303 PMid:3281000
- Messina
MF, Lombardo F, Meo A, Miceli M, Wasniewska M, Valenzise M, Ruggeri C,
Arrigo T, De Luca F. Three-year prospective evaluation of glucose
tolerance, beta-cell function and peripheral insulin sensitivity in
non-diabetic patients with thalassemia major. J Endocrinol Invest.
2002;25:497-501. https://doi.org/10.1007/BF03345490 PMid:12109619
- Jaruratanasirikul
S, Chareonmuang R, Wongcharnchailert M, Laosombat V, Sangsupavanich P,
Leetanaporn K. Prevalence of impaired glucose metabolism in
beta-thalassemic children receiving hypertransfusions with a suboptimal
dosage of iron-chelating therapy. Eur J Pediatr. 2008;167:873-876. https://doi.org/10.1007/s00431-007-0602-0 PMid:17899188
- Monge
L, Pinach S, Caramellino L, Bertero MT, Dall'omo A, Carta Q. The
possible role of autoimmunity in the pathogenesis of diabetes in
B-thalassemia major. Diabetes Metab. 2001;27:149-154.
- Kattamis
C, Ladis V, Tsoussis D, Kaloumenou I, Theodoridis C. Evolution of
glucose intolerance and diabetes in transfused patients with
thalassemia. Pediatr Endocrinol Rev. 2004;2 (Suppl 2):267-271.
- Crofts
C, Schofield G, Zinn C, Wheldon M, Kraft J. Identifying
hyperinsulinaemia in the absence of impaired glucose tolerance: an
examination of the kraft database. Diabetes Res Clin Pract.
2016;118:50-57. https://doi.org/10.1016/j.diabres.2016.06.007 PMid:27344544
- American
Diabetes Association. Classification and Diagnosis of Diabetes:
Standards of Medical Care in Diabetes - 2020. Diabetes Care. 2020;
43(Suppl.1): S14-S31. https://doi.org/10.2337/dc20-S002 PMid:31862745
- Bingley
PJ, Colman P, Eisenbarth GS, Jackson RA, McCulloch DK, Riley WJ, Gale
EA. Standardization of IVGTT to predict IDDM. Diabetes Care
1992;15:1313-1316. https://doi.org/10.2337/diacare.15.10.1313 PMid:1425095
- Vardi
P, Crisa L, Jackson RA. Predictive value of intravenous glucose
tolerance test insulin secretion less than or greater than the first
percentile in islet cell antibody positive relatives of type 1
(insulin-dependent) diabetic patients. Diabetologia.1991;34:93-102. https://doi.org/10.1007/BF00500379 PMid:2065854
- Fulwood
R, Johnson CL, Bryner JD. Hematological and nutritional biochemistry
reference data for persons 6 months-74 years of age: United States,
1976-1980. National Center for Health Statistics, Vital Health Stat
Series.1982; 11:1-173.
- De Sanctis V,
Elsedfy H, Soliman AT, Elhakim IZ, Kattamis C, Soliman NA, Elalaily R.
Clinical and Biochemical Data of Adult Thalassemia Major patients (TM)
with Multiple Endocrine Complications (MEC) versus TM Patients with
Normal Endocrine Functions: A long-term Retrospective Study (40 years)
in a Tertiary Care Center in Italy. Mediterr J Hematol Infect Dis. 2016
Apr 12;8(1):e2016022. https://doi.org/10.4084/mjhid.2016.022 PMid:27158435 PMCid:PMC4848017
- Matthews
DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC.
Homeostasis model assessment: insulin resistance and beta-cell function
from fasting plasma glucose and insulin concentrations in man.
Diabetologia. 1985; 28: 412-419. https://doi.org/10.1007/BF00280883 PMid:3899825
- Chen
H, Sullivan G, Quon MJ. Assessing the predictive accuracy of QUICKI as
a surrogate index for insulin sensitivity using a calibration model.
Diabetes. 2005;541914-1925. https://doi.org/10.2337/diabetes.54.7.1914 PMid:15983190
- Matsuda
M, De Fronzo RA. Insulin sensitivity indices obtained from oral glucose
tolerance testing: comparison with the euglycemic insulin clamp.
Diabetes Care.1999; 22:1462-1470. https://doi.org/10.2337/diacare.22.9.1462 PMid:10480510
- Płaczkowska
S, Pawlik-Sobecka L, Kokot I, Piwowar A. Estimation of reference
intervals of insulin resistance (HOMA), insulin sensitivity (Matsuda),
and insulin secretion sensitivity indices (ISSI-2) in Polish young
people. Ann Agric Environ Med. 2020;27:248-254. https://doi.org/10.26444/aaem/109225 PMid:32588601
- Retnakaran
R, Qi Y, Goran MI, Hamilton JK. Evaluation of proposed oral disposition
index measures in relation to the actual disposition index. Diabet Med.
2009; 26: 1198-203. https://doi.org/10.1111/j.1464-5491.2009.02841.x PMid:20002470
- Alder
R, Roesser EB. Introduction to probability and statistics.WH Freeman
and Company Eds. Sixth Edition.San Francisco (USA), 1975
- Petersmann
A, Müller-Wieland D, Müller UA, Landgraf R, Nauck M, Freckmann G,
Heinemann L, Schleicher E. Definition, Classification and Diagnosis of
Diabetes Mellitus. Exp Clin Endocrinol Diabetes. 2019;127:S1-S7. https://doi.org/10.1055/a-1018-9078 PMid:31860923
- De
Sanctis V, Soliman AT, Elsedfy H, Yaarubi SA, Skordis N, Khater D, El
Kholy M, Stoeva I, Fiscina B, Angastiniotis M, Daar S, Kattamis C. The
ICET-A Recommendations for the Diagnosis and Management of Disturbances
of Glucose Homeostasis in Thalassemia Major Patients. Mediterr J
Hematol Infect Dis. 2016 Oct 28;8(1):e2016058. https://doi.org/10.4084/mjhid.2016.058 PMid:27872738 PMCid:PMC5111521
- Iancu TC. Biological and ultrastructural aspects of iron overload: an overview. Pediatr Pathol. 1990;10:281-296. https://doi.org/10.3109/15513819009067114 PMid:2179920
- Iancu TC. Ultrastructural pathology of iron overload. Baillieres Clin Haematol. 1989;2:475-495. https://doi.org/10.1016/S0950-3536(89)80028-9
- Simpson
RJ, Deenmamode J, McKie AT, Raja KB, Salisbury JR, Iancu TC, Peters TJ.
Time-course of iron overload and biochemical, histopathological and
ultrastructural evidence of pancreatic damage in hypotransferrinaemic
mice. Clin Sci (Lond). 1997;93:453-462. https://doi.org/10.1042/cs0930453 PMid:9486091
- Suvarna
J, Ingle H, Deshmukh CT. Insulin resistance and beta cell function in
chronically transfused patients of thalassemia major. Indian Pediatr,
2006;43:393-400.
- Abdul-Ghani MA, DeFronzo RA. Pathogenesis of Insulin Resistance in Skeletal Muscle J Biomed Biotechnol.2010;2010:476279. https://doi.org/10.1155/2010/476279 PMid:20445742 PMCid:PMC2860140
- Santos
JL, Yevenes I, Cataldo LR, Morales M, Galgani J, Arancibia C, Vega J,
Olmos P, Flores M, Valderas JP, Pollak F. Development and assessment of
the disposition index based on the oral glucose tolerance test in
subjects with different glycaemic status. J Physiol Biochem.
2016;72:121-131. https://doi.org/10.1007/s13105-015-0458-0 PMid:26660757
- Singh B, Saxena A. Surrogate markers of insulin resistance: a review. World J Diabetes. 2010;1:36-47. https://doi.org/10.4239/wjd.v1.i2.36 PMid:21537426 PMCid:PMC3083884
- Gutch
M, Sukriti K, Razi SM, Gupta KK, Gupta A. Assessment of insulin
sensitivity/resistance. Indian J Endocrinol Metab. 2015;19:160-164. https://doi.org/10.4103/2230-8210.146874 PMid:25593845 PMCid:PMC4287763
- Hoffman
RP. Indices of insulin action calculated from fasting glucose and
insulin reflect hepatic, not peripheral, insulin sensitivity in
African-American and Caucasian adolescents. Pediatr Diabetes. 2008;
9:57-61. https://doi.org/10.1111/j.1399-5448.2007.00350.x PMid:18221434
- Karadas
N, Yurekli B, Bayraktaroglu S, Aydinok Y. Insulin secretion-sensitivity
index-2 could be a novel marker in the identification of the role of
pancreatic iron deposition on beta-cell function in thalassemia major.
Endocr J. 2019;66:1093-1099. https://doi.org/10.1507/endocrj.EJ19-0191 PMid:31527320
[TOP]