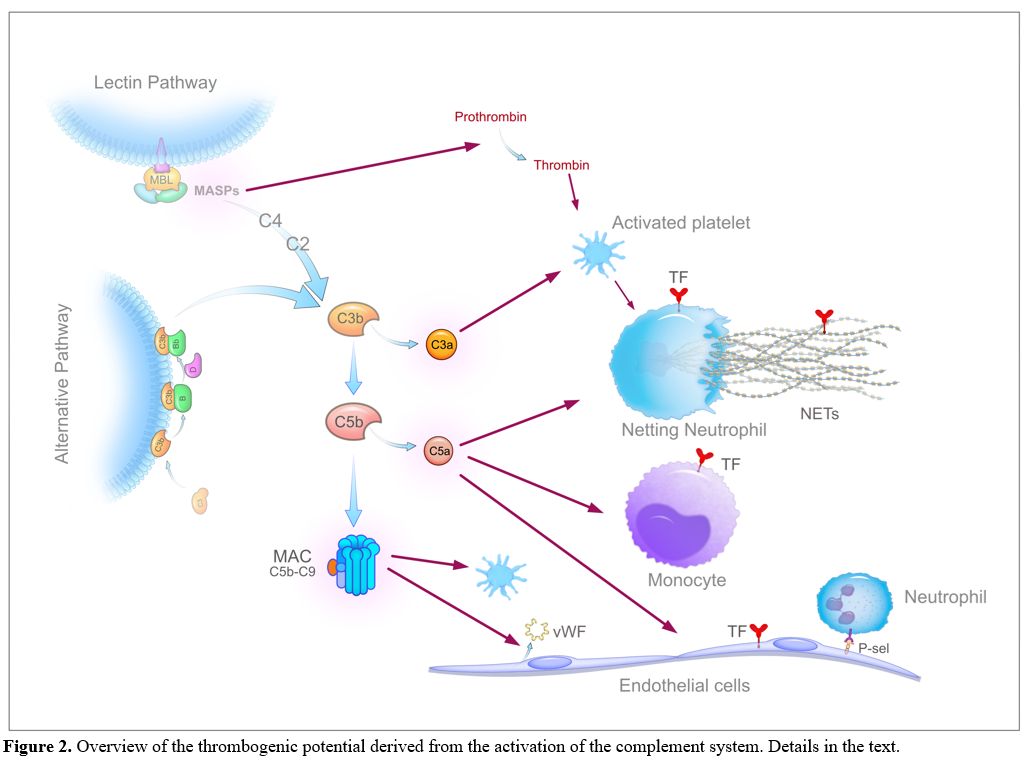
Given the close proximity to pneumocytes, alveolar macrophages are the first immune cells that recognize DAMPs and probably also the virus and/or its unique constituents (PAMPs, pathogen-associated molecular patterns) through specific receptors (PRRs, pattern recognition receptors, primarily the TLRs, Toll-like receptors), and respond with the synthesis and release of large amounts of proinflammatory mediators, mainly cytokines and chemokines. Cytokines secreted by alveolar macrophages, i.e., interleukin (IL)-6, IL-10 and tumour necrosis factor-alpha (TNF-α), have been detected in the lung by immunohistochemistry[53,54] and increased circulating levels of numerous cytokines, including IL-6, IL-8, IL-1β, and TNF-α, that correlate with disease severity have been reported in COVID-19 patients.[23,25,27,49] Moreover, SARS-Cov-2 activates the complement (C) alternative and lectin pathways through at least 2 surface proteins, i.e., the S protein, which, being heavily glycosylated with L-fucose and mannose, binds mannose-binding lectin (MBL), and the nucleocapsid protein (N protein), which binds MBL-associated serine protease 2 (MASP-2),[55,56] thus generating other potent proinflammatory mediators including the anaphylatoxins C3a and C5a, C3b and C5b-9 complex (membrane attack complex, MAC). The involvement of the complement system in COVID-19 is strongly supported by the extensive deposition of C5b-9, C4d, and MASP-2 within the lung microvasculature.[57] Strikingly, the SARS-CoV-2 S glycoprotein colocalized with C5b-9 and C4d in a subset of cases. In addition, patients with COVID-19 have elevated serum C5a and sC5b-9 levels.[58,59] All the generated inflammatory mediators then cause the recruitment into the alveolar and interstitial spaces of monocytes-macrophages and neutrophils, which are activated and, in their turn, release additional cytokines (the so-called "cytokine storm"), proteases and other mediators, such as reactive oxygen species (ROS), which work in concert to trigger a self-amplifying inflammatory cascade eventually resulting in a further injury of alveolar epithelial cells and hypoxic respiratory failure. In this setting, the vascular endothelium also plays a pivotal role. ACE2 is widely expressed on endothelial cells, and the presence of SARS-CoV-2 elements within these cells has been well documented.[60] Direct viral infection, combined with the milieu of proinflammatory mediators, will cause endothelial dysfunction/injury that strongly contributes to inflammation and tissue damage. Finally, platelets are activated by cytokines and complement products, as well as by the interaction with injured vessels and activated leukocytes, and release the content of their granules, including numerous proinflammatory mediators that further amplify the inflammatory reaction.[61,62] As expected, COVID-19 patients exhibit a marked elevation of acute-phase reactants (e.g., C-reactive protein, fibrinogen and ferritin). A schematic summary of the host inflammatory response to SARS-CoV-2 infection is reported in Figure 1.
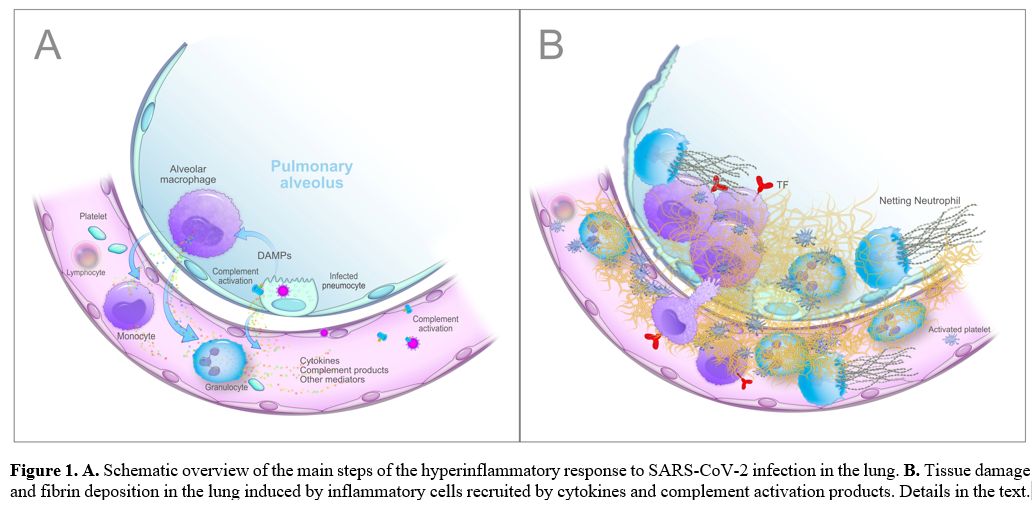 |
Figure
1. A. Schematic
overview of the main steps of the hyperinflammatory response to
SARS-CoV-2 infection in the lung. B. Tissue damage and fibrin
deposition in the lung induced by inflammatory cells recruited by
cytokines and complement activation products. Details in the text. |
In the light of these findings, a plausible hypothesis is that the plethora of inflammatory mediators generated during the persistent inflammatory status, along with the virus-derived components, drives thrombus formation in severe COVID-19 patients mainly through 1) the upregulation of the cellular procoagulant pathways, 2) the downregulation of physiological anticoagulant pathways and 3) the impairment of fibrinolysis. Virtually all cells participating in the inflammatory process, including lung epithelial cells, resident macrophages, monocytes, neutrophils, platelets, and endothelial cells, may variably contribute to these mechanisms.