Luca Guarnera1, Valentina Boldrini1, Gianmario Pasqualone1, Carolina Cimino2, Elisa Meddi1, Roberta Laureana1, Donata Trivigno2, Giovanni Del Poeta1, Alessandro Mauriello2, Lucia Anemona2, Massimiliano Postorino1 and Maria Cantonetti1.
1
Hematology, Department of Biomedicine and Prevention, University Tor Vergata, Rome, Italy.
2 Anatomic Pathology, Department of Biomedicine and Prevention, University Tor Vergata, Rome, Italy.
Correspondence to: Luca
Guarnera, Hematology, Department of Biomedicine and Prevention,
University Tor Vergata, Rome, Italy. Tel +39 0620908215, Fax +39
0620903246. E-mail:
lucaguarnera@live.com
Published: January 1, 2022
Received: September 11, 2021
Accepted: December 9, 2021
Mediterr J Hematol Infect Dis 2022, 14(1): e2022006 DOI
10.4084/MJHID.2022.006
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
T-cell
lymphomas and leukemias are highly heterogeneous groups of rare
disorders. We report a case of a 68-year-old man patient who developed
two different T-cell neoplasms (Large Granular Lymphocyte Leukemia
[LGLL] in 2018 and Peripheral T-cell non-Hodgkin lymphoma not otherwise
specified [PTCL-NOS] in 2019) with a previous diagnosis of B-cell
marginal zone lymphoma in 2010, treated with two lines of
chemo-immunotherapy. The coexistence of these different T-cell
neoplasms is rarely reported in the literature. Moreover, it is usually
described as an LGLL transformation into PTCL-NOS; differently from
these examples, herein, the simultaneous conditions appear to be driven
by different T-cell clones. Furthermore, the PTCL-NOS had quite unusual
behavior, with good disease control without intensive treatment.
Because of these features, it could belong to a subgroup of indolent
PTCL-NOS, not yet described in the WHO classification of T-cell
neoplasms, which could benefit from less aggressive treatment.
|
Introduction
Mature
T lymphoproliferative disorders are a spectrum of clinically and
biologically heterogeneous diseases. Pathogenesis is not fully
understood, although immune imbalance, intracellular signaling alterations, microenvironment, and
inflammatory stimuli seem to play a fundamental role. Primary
diagnostic sites can be solid tissues, lymph nodes, or peripheral blood
(PB).[1,2]
Large Granular Lymphocyte Leukemia (LGLL) is a rare
lymphoproliferative disorder usually diagnosed from PB, which can arise
from both T Cytotoxic Lymphocyte and NK cells. The first finding is
usually the identification of an increased number of circulating Large
Granular Lymphocytes (LGLs) that should be in number > 2 x 109/L (normal value: < 0.3 x 109/L),
even if several LGLL cases could present with an inferior lymphocyte
count; in this instance, the demonstration of clonality is mandatory
for diagnosis.[3,4]
In particular, T-LGLL sustained by the
proliferation of CD3+ T-LGLLs is the most frequent variant of the
disease.[5] Diagnosis requires the demonstration of clonal
lymphocytosis of LGLs (provided by T-cell receptor [TCR] gene
rearrangement analysis).[4]
A peculiar feature of the disease is
the association with autoimmune disorders and secondary neoplasms.
Isolated neutropenia (absolute neutrophil count [ANC] <1.50 x 109/L)
is the most common clinical condition, although low hemoglobin (Hb) and
platelets (Plts) levels may be found. Treatment is based on
immunosuppressive agents.[5,6]
Peripheral T-cell non-Hodgkin
lymphoma not otherwise specified (PTCL-NOS) is a broad category of
heterogeneous T cell diseases that cannot be further classified into
any other of the existing entities defined by the World Health
Organization classification (WHO).[7,8]
The diagnosis of PTCL-NOS
is based on typical histopathological features, an aberrant T-cell
immunophenotype, and a clonal TCR gene rearrangement. Although a
PTCL-NOS specific prognostic score has been proposed, the International
Prognostic Index (IPI) is still the recommended scoring system to
assess the prognosis in patients affected by this condition.[9]
Nowadays,
anthracycline-based regimens represent the standard first-line
treatment: the prognosis is generally poor, with a 5-year overall
survival (OS) of 20-30%, even if some authors suggest the existence of
an indolent PTCL variant with a good prognosis and possibility of
spontaneous regression.[7,10]
The case we report describes the
story of a patient with a previously diagnosed (and treated) marginal
zone B-cell lymphoma (MZL), who is currently affected by concomitant
T-LGLL and PTCL-NOS with indolent behaviour.
Case Report
The
story of our patient begins in 2010 when, at the age of 60, he was
referred to the hematology clinic because of cervical lymphadenopathy,
detection of mild anemia (Hb: 11.2 g /dL), and relative lymphocytosis
(ANC: 2.87 x 109/L, Ly: 3.71 x 109/L)
in routine complete blood count (CBC). Screening for hepatitis viruses,
HIV, EBV, CMV, Treponema Pallidum, Toxoplasma Gondii, autoimmunity
screening, and inflammatory markers were all negative. The
immunophenotypic profile of PB lymphocytes identified a clonal B cell
population. A whole-body computed tomography (CT) scan revealed several
lymphadenopathies (cervical, abdominal, pelvic localization); a
subsequent morphological and immunohistochemical study of bone marrow
(BM) showed medullary involvement by MZL. Considering the advanced
stage (Ann Arbor stage IV), the patient was started on R-CHOP
immuno-chemotherapy (Rituximab, Cyclophosphamide, Hydroxydaunorubicin,
Vincristine, Prednisone), achieving complete remission of the disease.
In April 2018, due to progressive neutropenia (ANC: 0,73 x 109/L)
and CT-documented recurrence of lymphadenopathies (mediastinal and deep
abdominal), he was reassessed with viral and autoimmunity screening,
then BM biopsy, that concluded for disease relapse. As a result,
second-line chemotherapy was started, according to R-Bendamustine
(Rituximab, Bendamustine) regimen.
The patient completed only four
cycles since hemolytic anemia (HA) occurred with Hb: 9.1 g/dL, reduced
haptoglobin (28 mg/dL), raised reticulocyte count (119 x 109/L; normal range: 30-110 x 109/L);
direct and indirect Coombs tests were negative, total bilirubin and
lactate dehydrogenase (LDH) levels resulted within normal ranges. HA
was managed with corticosteroid therapy (Prednisone 1 mg/Kg for three
weeks, then tapered to progressive reduction, so suspension). A
subsequent whole body re-evaluation CT scan documented a new complete
remission of the disease.
In September 2018, our patient was
hospitalized because of febrile neutropenia: at the admission, CBC
showed Hb: 12.9 g/dL, Plts: 82 x 109/L, ANC: 0.56 x 109/L,
Ly: 0.4, with LDH: 185 U/L. On suspicion of lymphoma recurrence, the
patient was reassessed by BM aspiration and biopsy, PB
immunophenotypic, and morphological examination. In addition, folates
and cobalamin levels, serologic and molecular essay for B and C
hepatitis, Epstein Barr virus, Parvovirus, Cytomegalovirus, and HIV
were investigated: all these tests were negative. A small interstitial
T cell infiltration (3%) was observed at BM biopsy, and
immunophenotypic analysis of marrow aspirate (lymphocyte gate)
highlighted a 10% of clonal cytotoxic T lymphocytes (CD3+, CD57+, CD8+,
CD2+, CD5+, CD7-, CD56+, CD4-, CD30-). Morphological examination
of PB smear revealed numerous LGLs (Figure 1):
the immunophenotypic profile of PB lymphocytes was equal to that found
in the BM aspirate (14% of total lymphocytes). Febrile neutropenia
resolved after two weeks of large spectrum antibiotic therapy.
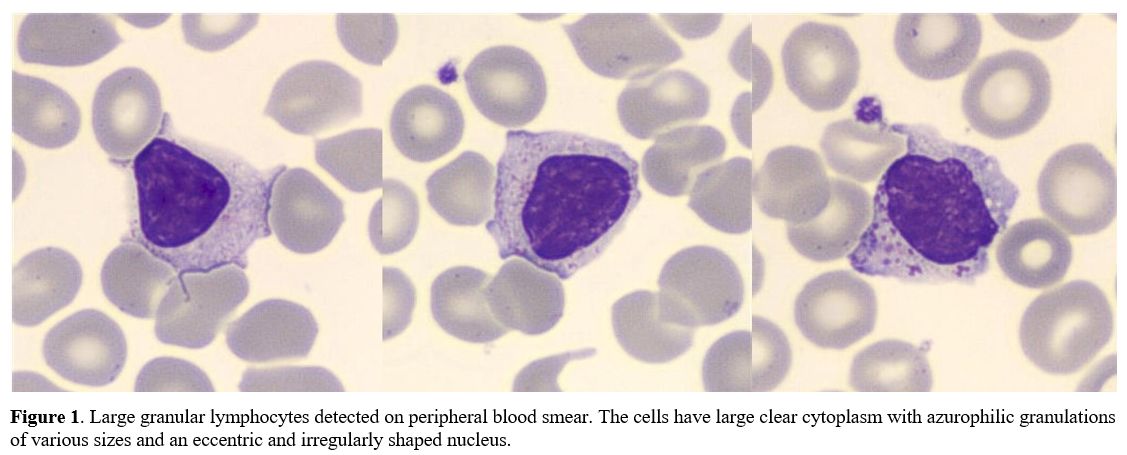 |
Figure
1. Large granular
lymphocytes detected on peripheral blood smear. The cells have large
clear cytoplasm with azurophilic granulations of various sizes and an
eccentric and irregularly shaped nucleus.
|
After
hematological recovery, subsequent TCR-γ polymerase chain reaction
showed a monoclonal expansion of the T-LGL population, with a diagnosis
of T-LGLL.
Considering the patient's general conditions (ECOG
grade 2) and the history of the previous two lines of chemotherapy, we
decided to start a closer clinical follow-up and to provide a support
therapy temporarily with granulocyte colony-stimulating factor (G-CSF)
(30 MUI weekly if ANC <500 x 109/L).
Unfortunately, as soon as the patient achieved good control of blood
count and satisfactory quality of life, he was lost at follow-up for
almost a year.
In October 2019, because of severe neutropenia, thrombocytopenia, mild anemia (Hb: 11 g/dL, Plts: 86.000 x 109/L, ANC: 0.49 x 109/L; Ly: 0.4 x 109/L),
itching, and LDH: 185 U/L, he was hospitalized and treated with
prophylactic antibiotic therapy and G-CSF support. Furthermore, he was
reassessed by viral and autoimmunity tests, CT scan, PB lymphocytes
immunophenotypic profile, BM aspiration, and BM biopsy. The whole-body
CT scan highlighted multiple lymphadenopathies (cervical, mediastinal,
deep abdominal, and pelvic; Ø max 27 mm in the deep abdomen).
Peripheral blood T cells immunophenotype (lymphocyte gate) confirmed an
11% of the original T-LGLL clone (CD3+, CD57+, CD8+, CD2+, CD5+, CD7-,
CD56+, CD4-, CD30-). Surprisingly, BM biopsy showed a medullary
infiltration by small T lymphocyte population different from that found
in September 2018: CD45+ bright, CD3+ bright, CD2+ bright, CD4+ bright,
CD27+, CD45RO+, TCR/αβ +, CD52+/-, CD8-, CD7-, CD30-, HLA-DR-, CD25-,
CD56-. The diagnosis was: medullary infiltration (20%) by PTCL-NOS (Figure 2), IPI score 3 (intermediate-high risk).
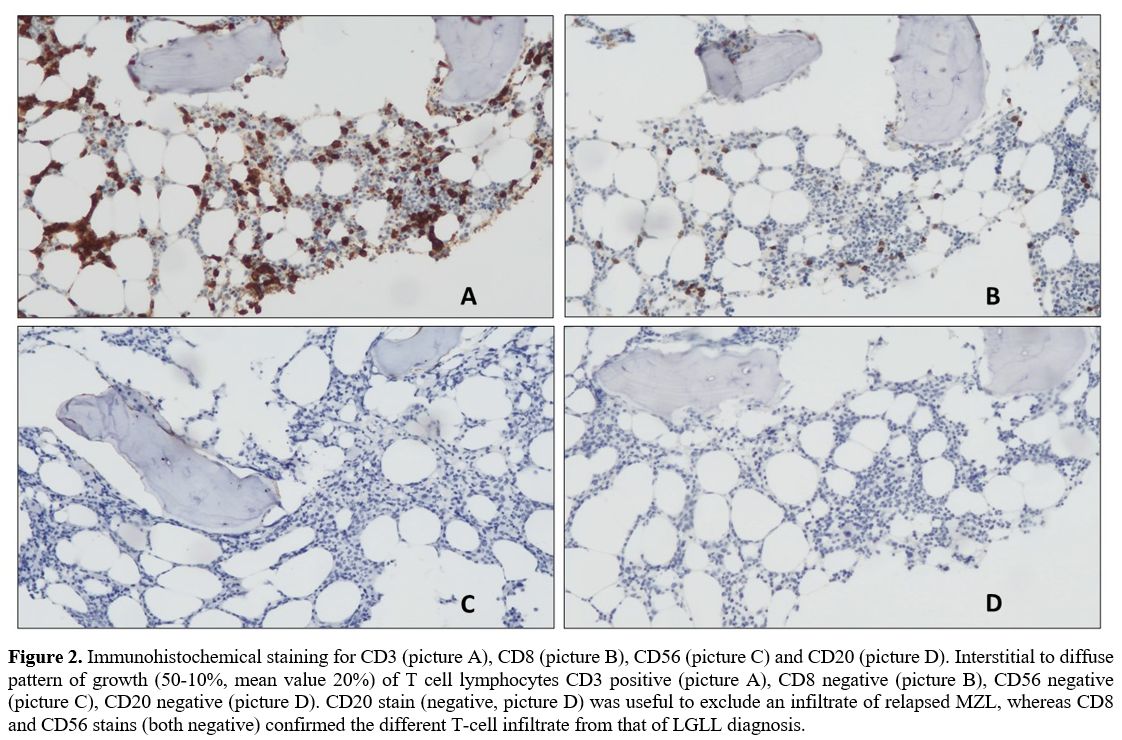 |
Figure 2. Immunohistochemical
staining for CD3 (picture A), CD8 (picture B), CD56 (picture C) and
CD20 (picture D). Interstitial to diffuse pattern of growth (50-10%,
mean value 20%) of T cell lymphocytes CD3 positive (picture A), CD8
negative (picture B), CD56 negative (picture C), CD20 negative (picture
D). CD20 stain (negative, picture D) was useful to exclude an
infiltrate of relapsed MZL, whereas CD8 and CD56 stains (both negative)
confirmed the different T-cell infiltrate from that of LGLL diagnosis.
|
Due
to the patient's poor condition (ECOG grade 3), he was started on
corticosteroid therapy (Prednisone 25 mg/die) as provisional
bridge-therapy to evaluate the most appropriate approach to managing
the dual condition.
Normalization of blood counts and
improvement of itchy symptoms were achieved in a few weeks, and
corticosteroid therapy was progressively taped to 7,5 mg/die. The
patient then refused to undergo a new intravenous chemotherapy regimen
and continued to receive corticosteroids as palliative treatment.
Corticosteroid
therapy was definitively interrupted in May 2020 since bilateral
aseptic osteonecrosis of the femoral head occurred. However, after
surgical intervention for bilateral total hip arthroplasty, we decided
to start therapy with antifolates drug (subcutaneous methotrexate – MTX
- 10 mg/m2/week), with good control of symptoms and blood count until February 2021.
Discussion and Conclusions
Immune system impairment has been linked to lymphoproliferative disorders arising from B and less frequently by T cells.[11]
We
described the case of a patient with immune system impairment (previous
MZL treated with two lines of chemo-immunotherapy) who developed two
T-cell neoplasms: firstly LGLL, then PTCL-NOS. The short time interval
between MZL relapse and LGLL diagnosis could lead to hypothesize a
reciprocal relationship. The correlation between these two conditions
is described in the literature,[12] and several hypotheses have been formulated. Goyal et al. and Viny et al.[12,13]
agree on two main possible mechanisms: a common antigenic trigger or a
humoral stimulus serving as a lymphocyte expansion supporter. However,
B and T cell populations are unlikely to be clonally related.
On the other hand, the two T-cell lymphoproliferative disorders herein presented are only described in few reports,[14-16]
but authors always refer to a "transformation" of the previous T-LGLL
into a T-cell lymphoma; furthermore, all these cases describe an
aggressive behavior of the secondary lymphoma. The clinical
behavior of our patient's PTCL NOS is quite unusual and could be framed
as a particular subgroup in the subset of T-cell lymphomas, currently
not described in WHO classification of T-cell neoplasms.[8]
Our patient achieved a good control of disease, in line with the
experiences of other authors, who reported stable disease or long-term
survival even without intensive treatment.
Even if it seems difficult to distinguish indolent PTCL-NOS at diagnosis, Hayashi et al.[17]
described some characteristic features of these entities: regardless of
nodal or extranodal involvement, lymphoid cells are typically small in
size, with oval or slightly irregular nuclei and pale cytoplasm. Of
note: all the described cases showed a Ki67 index <10%.[10,17]
Unfortunately,
this marker was not investigated in our patient at the time of PTCL-NOS
diagnosis and subsequently was not carried out due to insufficient
material.
Ki67 index seems to represent a reliable tool and,
together with a precise morphological and genetic-molecular
classification, could help define this entity better.
In this
respect, it is essential to understand how to distinguish aggressive
versus indolent PTCL-NOS to avoid overtreatment of these conditions and
plan a proper follow-up.
References
- Jevremovic D, Olteanu H. Flow Cytometry
Applications in the Diagnosis of T/NK-Cell Lymphoproliferative
Disorders. Cytometry Part B - Clinical Cytometry 2019. https://doi.org/10.1002/cyto.b.21768 PMid:30729667
- Pizzi
M, Margolskee E, Inghirami G. Pathogenesis of Peripheral T Cell
Lymphoma. Annual Review of Pathology: Mechanisms of Disease 2018. https://doi.org/10.1146/annurev-pathol-020117-043821 PMid:29414251
- Semenzato
G, Zambello R, Starkebaum G, Oshimi K, Loughran TP. The
lymphoproliferative disease of granular lymphocytes: Updated criteria
for diagnosis. Blood 1997. https://doi.org/10.1182/blood.V89.1.256 PMid:8978299
- Lamy T, Moignet A, Loughran TP. LGL leukemia: From pathogenesis to treatment. Blood 2017. https://doi.org/10.1182/blood-2016-08-692590 PMid:28115367
- Barilà
G, Calabretto G, Teramo A, Vicenzetto C, Gasparini VR, Semenzato G, et
al. T cell large granular lymphocyte leukemia and chronic NK
lymphocytosis. Best Practice and Research: Clinical Haematology 2019. https://doi.org/10.1016/j.beha.2019.06.006 PMid:31585621
- Teramo
A, Barilà G, Calabretto G, Ercolin C, Lamy T, Moignet A, et al. STAT3
mutation impacts biological and clinical features of T-LGL leukemia.
Oncotarget 2017. https://doi.org/10.18632/oncotarget.18711 PMid:28977911 PMCid:PMC5617471
- Broccoli A, Zinzani PL. Peripheral T-cell lymphoma, not otherwise specified. Blood 2017. https://doi.org/10.1182/blood-2016-08-692566 PMid:28115372
- Swerdlow
SH, Campo E, Pileri SA, Lee Harris N, Stein H, Siebert R, et al. The
2016 revision of the World Health Organization classification of
lymphoid neoplasms. Blood 2016. https://doi.org/10.1182/blood-2016-01-643569 PMid:26980727 PMCid:PMC4874220
- d'Amore
F, Gaulard P, Trümper L, Corradini P, Kim WS, Specht L, et al.
Peripheral T-cell lymphomas: ESMO Clinical Practice Guidelines for
diagnosis, treatment and follow-up. Annals of Oncology 2015. https://doi.org/10.1093/annonc/mdv201 PMid:26314772
- Lee
J, Park K, Kim KH, Bang HI, Yoon SY, Choi IH. Diagnostic challenges of
indolent peripheral T cell lymphoma: A case report and literature
review. Medicine 2020. https://doi.org/10.1097/MD.0000000000022657 PMid:33080706 PMCid:PMC7571990
- Nijland
ML, Koens L, Pals ST, Ten Berge IJM, Bemelman FJ, Kersten MJ.
Clinicopathological characteristics of T-cell non-Hodgkin lymphoma
arising in patients with immunodeficiencies: A single-center case
series of 25 patients and a review of the literature. Haematologica
2018. https://doi.org/10.3324/haematol.2017.169987 PMid:29269521 PMCid:PMC5830383
- Goyal
T, Thakral B, Wang SA, Bueso-Ramos CE, Shi M, Jevremovic D, et al.
T-Cell Large Granular Lymphocytic Leukemia and Coexisting B-Cell
Lymphomas. American Journal of Clinical Pathology 2018. https://doi.org/10.1093/ajcp/aqx146 PMid:29365010
- Viny
A, Lichtin A, Pohlman B, Loughran T, Maciejewski J. Chronic B-cell
dyscrasias are an important clinical feature of T-LGL leukemia.
Leukemia and Lymphoma 2008. https://doi.org/10.1080/10428190801932635 PMid:18452068
- Matutes
E, Wotherspoon AC, Parker NE, Osuji N, Isaacson PG, Catovsky D.
Transformation of T-cell large granular lymphocyte leukaemia into a
high-grade large T-cell lymphoma. British Journal of Haematology 2001. https://doi.org/10.1046/j.1365-2141.2001.03220.x PMid:11843812
- Belhadj
M, Mansour D, Kaltenbach S, Deau-Fischer B, Franchi P, Tamburini J, et
al. T-cell large granular lymphocyte leukemia transfomation into
aggressive t-cell lymphoma: A report of two cases with molecular
characterization. Haematologica 2019. https://doi.org/10.3324/haematol.2018.205542 PMid:30573508 PMCid:PMC6395332
- Nakmaura
N, Carreras J, Kikuti YY, Miyaoka M, Tomita S, Ishida F. RICHTER
TRANSFORMATION OF T-CELL LARGE GRANULAR CELL LEUKEMIA. Hematological
Oncology 2017. https://doi.org/10.1002/hon.2439_164
- Hayashi
E, Takata K, Sato Y, Tashiro Y, Tachiyama Y, Sawada-Kitamura S, et al.
Distinct morphologic, phenotypic, and clinical-course characteristics
of indolent peripheral T-cell lymphoma. Human Pathology 2013. https://doi.org/10.1016/j.humpath.2013.03.002 PMid:23706909
[TOP]