Beatrice Borsellino1,2, Arianna Savi1, Maria Rosaria Pascale1, Elisa Meddi1, Maria Ilaria Del Principe1, Antonio Cristiano1, Tiziana Ottone1,3, Maria Cristina Rapanotti1,5, Mariadomenica Divona1,4, Serena Travaglini1, Enrico Attardi1, Raffaele Palmieri1, Elisa Buzzatti1, Francesco Buccisano1 and Maria Teresa Voso1,3.
1
Department of Biomedicine and Prevention, University of Tor Vergata, Rome, Italy.
2
Hematology Clinic, Department of Clinical and Molecular Sciences,
DISCLIMO, AOU Ospedali Riuniti-Università Politecnica delle Marche,
Ancona, Italy.
3 Neuro-Oncohematology Unit, IRCCS Fondazione Santa Lucia, Rome, Italy.
4 UniCamillus‐Saint Camillus International University of Health Sciences, Rome Italy.
5 Department of Experimental Medicine, Tor Vergata University of Rome, Rome, Italy.
Correspondence to:
Prof.ssa Maria Teresa Voso. Department of Biomedicine and Prevention.
Tor Vergata University. Rome, Italy.
Published: July 1, 2022
Received: April 7, 2022
Accepted: June 18, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022058 DOI
10.4084/MJHID.2022.058
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
evolution of myeloproliferative neoplasms (MPN) to acute myeloid
leukemia (AML) occurs in 2-10% of patients, depending on the MPN
subtype, treatment, and follow-up length. The reverse-path from AML to
MPN has been rarely reported.
We herein present a 75 years old woman with AML, in whom a JAK2-V617F positive polycythemia vera (PV) emerged during follow-up, 19 months from the end of consolidation treatment.
JAK2-V617F mutation screening retrospectively performed by Next Generation Sequencing (NGS) and JAK2
MutaScreen was negative on the bone marrow sample collected at AML
diagnosis. However, using digital droplet PCR (ddPCR), we detected a
minor JAK2
V617F mutated clone at AML onset. In addition, a TET2 R550 mutated
clone persisted at stable levels throughout the disease course.
This
case shows that a very small MPN clone masked at AML diagnosis may
expand after treatment end and be erroneously interpreted as MPN
evolving from AML. Very sensitive techniques such as ddPCR may help to
unravel the true disease history in these cases.
|
Introduction
Myeloproliferative
neoplasms (MPNs) are acquired clonal disorders characterized by
aberrant hematopoietic proliferation and the increased tendency towards
leukemic transformation.[1,2] The risk of leukemic
progression varies depending on the MPN subtype, with primary
myelofibrosis (PMF) associated with the highest transformation risk
(10-year risk: 10-20%), followed by polycythemia vera (PV) and
essential thrombocythemia (TE) (10-year risk: 2-4% and about 1%,
respectively).[3-5] Activating mutations of the JAK/STAT pathway, primarily in the JAK2 gene (mainly JAK2
V617F), are frequently found in patients with MPNs and rarely occur in
de novo Acute Myeloid Leukemia (AML) with a frequency of about 1%.[1,6,7] This highlights the crucial role of JAK2 mutations as phenotypic drivers in MPN, particularly in PV, where JAK2 V617F mutation is found in 95% of patients.[2]
Here, we used this molecular marker to trace the origin of the disease
in a case of PV manifested two years after AML diagnosis.
PV is
a MPN characterized by abnormal red blood precursor cells proliferation
and erythrocytosis, often associated with thrombocytosis and
leukocytosis. Despite progression to AML is a possible evolution of
MPN, only a few cases of JAK2 V617F-positive PV developing while in long-term remission from AML have been previously described.[8-12]
Here,
we reported on a patient diagnosed with AML, who was treated with
conventional 7+3 based-chemotherapy, achieved complete remission, and
developed a JAK2-mutated
PV two years after the end of consolidation treatment. We were
interested in the biological features of the two diseases to define
better the onset of the MPN clone and its kinetics.
A 73-year-old fit female came to our observation in June 2019 due to mild anemia (Hb 10.6 g/dl), monocytosis (1.760 x109/L), and neutropenia (0.700 x109/L).
Personal and familiar history was negative, and blood counts assessed
two years before were normal. The peripheral blood smear showed 25%
myeloid blasts, and the bone marrow aspirate contained 80%
CD34+/CD33+/CD7+, CD117+, CD13+, DR+, MPO+ blasts, consistent with the
diagnosis of AML. Cytogenetics showed monosomy of chromosome X in 6 of
20 metaphases, while the molecular profile was negative for recurrent
mutations, including mutations in FLT3, NPM1, IDH1, and IDH2
genes. Next-generation sequencing (NGS) was performed on DNA extracted
from bone marrow mononuclear cells (BM-MNC) and highlighted the
presence of TET2 p.Y1560* (variant allele frequency, VAF: 39.3%) and TET2 p.R550* (VAF: 44.9%) mutations. According to ELN 2017 risk stratification, the disease was classified as intermediate risk.[13]
She started the "7+3" chemotherapy regimen, obtaining complete
remission (CR) and minimal residual disease negativity (MRD) by flow
cytometry (sensitivity < 0.035%). She then underwent two high-dose
cytarabine consolidation courses. Treatment was complicated by one
episode of pulmonary embolism during induction and later on by a
catheter-related thrombosis during consolidation treatment.
Thrombophilia tests, including antithrombin deficiency, protein C and S
deficiency, factor V Leiden, prothrombin mutation, dysfibrinogenemia,
anticardiolipin antibodies, anti-beta2 glycoprotein I antibodies,
hyperhomocysteinemia and lupus anticoagulant, were negative. She was
then monitored through sequential complete blood counts and BM
aspirates, performed every three months. After two years, while still
in CR, the patient's blood counts showed a progressive increase in
hemoglobin and hematocrit (Figure 1). Erythropoietin levels were within normal lower limits (3.7 mU/mL, normal range: 3.7-31.5), and the JAK2-V617F
mutation was positive in peripheral blood. The bone marrow biopsy
showed increased cellularity (30%), normal myeloid maturation, less
than 3% CD34+ blast cells, and no signs of fibrosis (MF-0, according to
2017 WHO classification),[14] thus confirming the diagnosis of PV. The NGS analysis showed the following mutations: TET2 p.R550*(VAF: 46%), and JAK2 p.V617F (VAF: 43.4%), while cytogenetics was normal (46, XX). Figure 2 shows the expansion of the JAK2
V617F mutated clone. Due to the increased hematocrit, the patient
started therapeutic phlebotomies and hydroxyurea, according to the
"high" thrombotic risk category.[15]
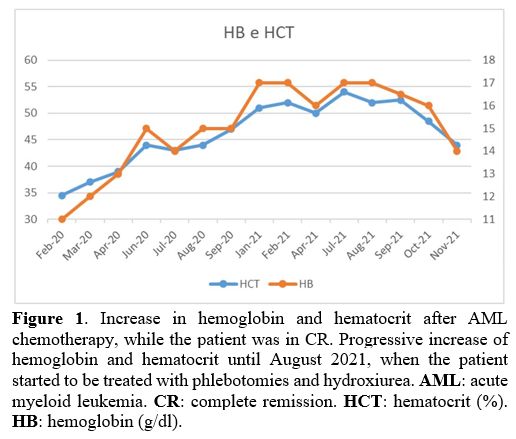 |
Figure 1. Increase in
hemoglobin and hematocrit after AML chemotherapy, while the patient was
in CR. Progressive increase of hemoglobin and hematocrit until August
2021, when the patient started to be treated with phlebotomies and
hydroxiurea. AML: acute myeloid leukemia. CR: complete remission. HCT: hematocrit (%). HB: hemoglobin (g/dl). |
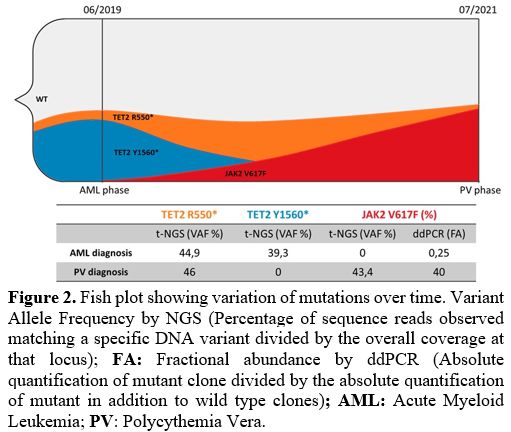 |
Figure 2. Fish plot
showing variation of mutations over time. Variant Allele Frequency by
NGS (Percentage of sequence reads observed matching a specific DNA
variant divided by the overall coverage at that locus); FA:
Fractional abundance by ddPCR (Absolute quantification of mutant clone
divided by the absolute quantification of mutant in addition to wild
type clones); AML: Acute Myeloid Leukemia; PV: Polycythemia Vera. |
We then traced back the JAK2
V617F mutated clone at the time of AML diagnosis using the MutaScreen
assay (Ipsogen, Luminy Biotech, Marseille, France), which provides 2%
cut-off sample (COS) positivity, and NGS (2% sensitivity). Both were
negative for JAK2 mutation. To further exclude the presence of a JAK2-mutated
clone at the time of AML diagnosis, we performed a digital droplet PCR
(ddPCR) assay, which has a sensitivity of 0.05%,[16,17] which resulted positive for JAK2 V617F (Figure 3),
suggesting that this mutation was present at a subclonal levels at the
time of AML diagnosis and undetectable when measured using other
conventional diagnostic tools.
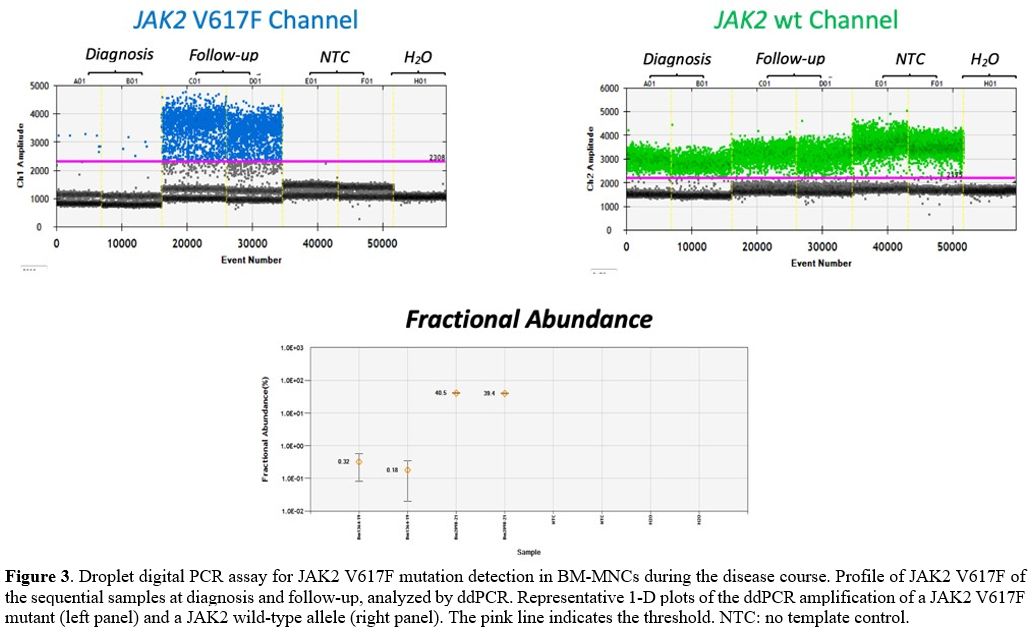 |
Figure 3. Droplet digital
PCR assay for JAK2 V617F mutation detection in BM-MNCs during the
disease course. Profile of JAK2 V617F of the sequential samples at
diagnosis and follow-up, analyzed by ddPCR. Representative 1-D plots of
the ddPCR amplification of a JAK2 V617F mutant (left panel) and a JAK2
wild-type allele (right panel). The pink line indicates the
threshold.NTC: no template control.
|
Discussion
We
report on a patient erroneously diagnosed as MPN evolving from AML, in
which a small mutated clone present at the time of AML onset expanded
during AML follow-up and manifested as overt PV. Indeed, during AML
follow-up, the high levels of hemoglobin and the persistently increased
hematocrit provided the clues to research the JAK2 mutation in the peripheral blood, which resulted positive.
Tracking back the JAK2
mutation at the AML diagnosis, we found a minor subclone detectable by
ddPCR only, confirming this approach's ability to detect mutant cases
early during the disease course. The small clone was probably
suppressed by the overt AML blast infiltration at diagnosis. Now the
question is whether the two diseases are independent, or can AML be
considered an evolution of PV in this case?
The patient was found to carry two TET2
mutations at high VAF. One disappeared at the time of achievement of
CR, indicating that it was probably related to the AML clone, while the
other was present at high VAF both at AML and PV diagnosis, suggesting
that this was a large mutated clone, generating genomic instability,
hence the predisposition for the development of both diseases.[18]
MPN
may have arisen independently, but it is impossible to exclude that the
diseases might have evolved from a common precursor.
The second hypothesis would be that AML represented the evolution of MPN. JAK2-WT development of PV has been described in 2-4% of cases, and usually, in these cases, TET2-mutation occurs "first", as in our patient.[5]
However, secondary AMLs are generally characterized by poor prognosis
and unfavorable or complex karyotype. In contrast, in this case, the
intermediate-risk and absence of adverse mutations like TP53 render the
diagnosis of de novo AML more likely. Accordingly, complete remission
was achieved after "7+3" induction chemotherapy followed by a prolonged
disease-free survival, which has now reached 33 months.[19]
The
two diseases are most likely independent. Indeed, Hb, HCT, WBC, and
PLTs measured two years before AML diagnosis were normal, indicating
that MPN was not present then. The patient did not have any other blood
tests performed until AML diagnosis; however, it is unlikely that a MPN
could have evolved in AML during this time, given that the average
evolution time is usually longer (incidence of leukemic transformation
of PV and ET: 2%-5% at 15 years).[20] The patient had
a pulmonary embolism and a catheter-related thrombosis during AML
chemotherapy. Thrombophilic and cardiovascular risk factors (obesity,
hypercholesterolemia, hypertension, smoking, and second tumor) were
negative. Therefore, we can suppose that the thrombotic tendency could
be favored by the presence of the small myeloproliferative clone
present at AML diagnosis.
One additional hypothesis is that the initial low-level JAK2
mutation was part of clonal hematopoiesis of indeterminate potential
(CHIP), where JAK2 is one of the most frequently mutated genes.[21,22,12]
In
conclusion, we interpret the two diseases as simultaneous and not
sequential, further supporting the idea of competing clones in myeloid
malignancies.[23] The lack of sequential samples after AML diagnosis did not allow for the correlation of the kinetics of emergence of the JAK2
mutant clone. Previous studies reported in the literature speculated
the possibility that intensive induction chemotherapy for AML, which
results in the ablation of the BM microenvironment, may provide a
suitable niche for pre-existing JAK2 V617F-positive stem cells with clonal potential to expand, resulting in the appearance of PV.[8-12]
In
summary, our report indicates that the coexistence of AML and MPNs
could be possible beyond the natural history of myeloid malignancies,
leading physicians to proceed to the diagnostic algorithm of MPN also
in the presence of subtle clues that could suggest a "color change"
towards myeloproliferative phenotype. The availability of sensitive
diagnostic techniques such as ddPCR may provide the necessary
diagnostic support to unravel these situations.
Acknowledgements
This
study was supported in part by AIRC 5x1000 call "Metastatic disease:
the key unmet need in oncology" to MYNERVA project, #21267 (MYeloid
NEoplasms Research Venture) AIRC. A detailed description of the MYNERVA
project is available at http://www.progettoagimm.it), by Ricerca finalizzata, code NET-2018-12365935 and PRIN grant N. 2017WXR7ZT to MTV.
References
- Aynardi J, Manur R, Hess PR, Chekol S, Morrissette
JJD, Babushok D et al. JAK2 V617F-positive acute myeloid leukaemia
(AML): a comparison between de novo AML and secondary AML transformed
from an underlying myeloproliferative neoplasm. A study from the Bone
Marrow Pathology Group. Br J Haematol 2018; 182: 78-85.
https://doi.org/10.1111/bjh.15276 PMid:29767839
- Passamonti
F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L et al. A prospective
study of 338 patients with polycythemia vera: the impact of JAK2
(V617F) allele burden and leukocytosis on fibrotic or leukemic disease
transformation and vascular complications. Leukemia 2010; 24:
1574-1579. https://doi.org/10.1038/leu.2010.148 PMid:20631743
- Mesa
RA, Verstovsek S, Cervantes F, Barosi G, Reilly JT, Dupriez B et al.
Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis
(post-PV MF), post essential thrombocythemia myelofibrosis (post-ET
MF), blast phase PMF (PMF-BP): Consensus on terminology by the
international working group for myelofibrosis research and treatment
(IWG-MRT). Leuk Res 2007; 31: 737-740.
https://doi.org/10.1016/j.leukres.2006.12.002 PMid:17210175
- Tefferi
A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L et al.
Long-term survival and blast transformation in molecularly annotated
essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood
2014; 124: 2507-13; quiz 2615.
https://doi.org/10.1182/blood-2014-05-579136 PMid:25037629
PMCid:PMC4199952
- Dunbar AJ, Rampal RK, Levine R. Leukemia
secondary to myeloproliferative neoplasms. Blood. 2020;136(1):61-70.
https://doi.org/10.1182/blood.2019000943 PMid:32430500 PMCid:PMC7332899
- Theocharides A, Boissinot M, Girodon F, Garand R, Teo S-S,
Lippert E et al. Leukemic blasts in transformed JAK2-V617F-positive
myeloproliferative disorders are frequently negative for the JAK2-V617F
mutation. Blood 2007; 110: 375-379.
https://doi.org/10.1182/blood-2006-12-062125 PMid:17363731
- Kralovics
R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR et al. A
gain-of-function mutation of JAK2 in myeloproliferative disorders. N
Engl J Med 2005; 352: 1779-1790. https://doi.org/10.1056/NEJMoa051113
PMid:15858187
- Youk H-J, Cho C-H, Lee J-H, Choi CW, Lim CS,
Yoon S-Y. A rare case of polycythemia vera following acute
undifferentiated leukemia remission. Ann. Lab. Med. 2014; 34: 469-470.
https://doi.org/10.3343/alm.2014.34.6.469 PMid:25368824
PMCid:PMC4215407
- Portell CA, Sekeres MA, Rogers HJ, Tiu R
V. De novo polycythaemia vera arising 5 years following acute myeloid
leukemia remission: suggestion of a chemotherapy resistant JAK2 clone.
Br. J. Haematol. 2012; 157: 266-267. https://doi.org/10.1111/j.1365-2141.2011.08972.x PMid:22150289
- Antonioli
E, Guglielmelli P, Poli G, Santini V, Bosi A, Vannucchi AM.
Polycythemia vera following autologous transplantation for AML:
insights on the kinetics of JAK2V617F clonal dominance. Blood 2007;
110: 4620-4621. https://doi.org/10.1182/blood-2007-07-103267
PMid:18056850
- Belotti A, Doni E, Elli E, Rossi V, Pioltelli P,
Pogliani EM. Development of polycythemia vera after
chemotherapy-induced remission of acute myeloid leukemia: a case
report. Acta Haematol. 2011; 126: 52-3.
https://doi.org/10.1159/000324468 PMid:21454967
- Langabeer
SE, O'Flynn DW, Cahill MR. Polycythemia vera emerging eighteen years
after acute myeloid leukemia diagnosis. Blood Res. 2021; 56: 121-123.
https://doi.org/10.5045/br.2021.2021040 PMid:33986187 PMCid:PMC8246042
- Döhner
H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Ebert BL et al.
Diagnosis and management ofAML in adults: 2017 ELN recommendations from
an international expert panel. Blood 2017; 129: 424-448.
https://doi.org/10.1182/blood-2016-08-733196 PMid:27895058
PMCid:PMC5291965
- Barbui T, Thiele J, Gisslinger H,
Kvasnicka HM, Vannucchi AM, Guglielmelli P et al. The 2016 WHO
classification and diagnostic criteria for myeloproliferative
neoplasms: document summary and in-depth discussion. Blood Cancer J
2018; 8: 15. https://doi.org/10.1038/s41408-018-0054-y PMid:29426921
PMCid:PMC5807384
- Tefferi A, Barbui T. Polycythemia vera and
essential thrombocythemia: 2021 update on diagnosis,
risk-stratification and management. Am J Hematol 2020; 95: 1599-1613.
https://doi.org/10.1002/ajh.26008 PMid:32974939
- Waterhouse
M, Follo M, Pfeifer D, von Bubnoff N, Duyster J, Bertz H et al.
Sensitive and accurate quantification of JAK2 V617F mutation in chronic
myeloproliferative neoplasms by droplet digital PCR. Ann Hematol 2016;
95: 739-744. https://doi.org/10.1007/s00277-016-2623-0 PMid:26931113
- Krichevsky
S, Prus E, Perlman R, Fibach E, Ben-Yehuda D. The JAK2V617F mutation in
normal individuals takes place in differentiating cells. Blood Cells
Mol Dis 2017; 63: 45-51. https://doi.org/10.1016/j.bcmd.2017.01.001 PMid:28126623
- Sallman
DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA et
al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant
Myelodysplastic Syndromes. J Clin Oncol Off J Am Soc Clin Oncol 2021;
39: 1584-1594. https://doi.org/10.1200/JCO.20.02341 PMid:33449813
PMCid:PMC8099410
- Grinfeld J, Nangalia J, Baxter EJ, Wedge DC,
Angelopoulos N, Cantrill R et al. Classification and Personalized
Prognosis in Myeloproliferative Neoplasms. N Engl J Med 2018; 379:
1416-1430. https://doi.org/10.1056/NEJMoa1716614 PMid:30304655
PMCid:PMC7030948
- Luque Paz D, Jouanneau-Courville R, Riou
J, Ianotto J-C, Boyer F, Chauveau A et al. Leukemic evolution of
polycythemia vera and essential thrombocythemia: genomic profiles
predict time to transformation. Blood Adv 2020; 4: 4887-4897.
https://doi.org/10.1182/bloodadvances.2020002271 PMid:33035330
PMCid:PMC7556129
- Jaiswal S, Fontanillas P, Flannick J, Manning
A, Grauman PV, Mar BG et al. Age-related clonal hematopoiesis
associated with adverse outcomes. N Engl J Med. 2014; 371: 2488-98.
https://doi.org/10.1056/NEJMoa1408617 PMid:25426837 PMCid:PMC4306669
- Desai
P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, Samuel M
et al. Somatic mutations precede acute myeloid leukemia years before
diagnosis. Nat Med. 2018; 24: 1015-1023.
https://doi.org/10.1038/s41591-018-0081-z PMid:29988143
PMCid:PMC6849383
- Al-Kali A, Verstovsek S, Kantarjian H, Luthra
R, Cortes J. Competing cell clones in myeloproliferative neoplasm.
Blood. 2010; 116: 5074-5075.
https://doi.org/10.1182/blood-2010-05-284885 PMid:21127188
PMCid:PMC4916558
[TOP]