
Review Articles
Vol. 1 No. 1 (2009): Progress in Diagnosis and Therapy of Thalassemia and Hemoglobinopathies
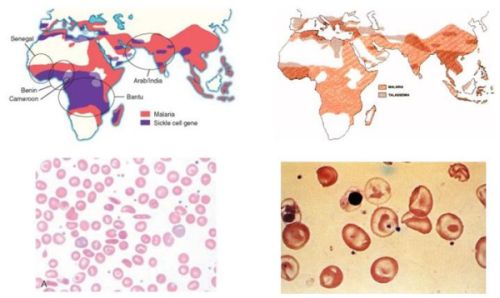
PATHOPHISIOLOGY OF SICKLE CELL DISEASE AND NEW DRUGS FOR THE TREATMENT
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Published: December 26, 2009
12155
Views
655
Downloads
11084
HTML
Hematology




