Received: July 11, 2013
Accepted: November 22, 2013
Meditter J Hematol Infect Dis 2014, 6(1): e2014001, DOI 10.4084/MJHID.2014.001
This article is available on PDF format at:

Darcie Deaver, MS,1 Mojdeh Naghashpour, MD, PhD,2 and Lubomir Sokol, MD, PhD1
1 Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL
2 Department of Hematopathology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Kikuchi-Fujimoto Disease (KFD), also known as histiocytic lymphadenitis, is a benign, self-limiting disease that manifests primarily as cervical lymphadenopathy but may include low-grade fever, headache, and fatigue. There is a higher incidence of KFD in women aged 20-35 years and in Asian populations. A PubMed search revealed 590 articles that described KFD. Of these, 22 cases have been fully described in the United States. Ten of the 22 (45%) patients were male and 12 (55%) were female, with 20% Caucasian, 20% Asian American, and the remaining 60% of other ethnic backgrounds. In this study, we describe an additional 3 cases of KFD and discuss the diagnosis, pathology, and management of KFD. |
Introduction
Kikuchi-Fujimoto
Disease (KFD), also known as histiocytic necrotizing lymphadenitis, was
first described independently in 1972 by Kikuchi and Fujimoto.[1,2] The initial presentation of KFD includes cervical lymphadenopathy, low-grade fever, headache, and fatigue.[3,4]
Other less common symptoms include nausea, vomiting, night sweats, and
weight loss. Cutaneous manifestations may also be a presenting sign;
however, this is a less common finding.[4] Laboratory studies may reveal leukopenia, anemia, elevated erythrocyte sedimentation rate, and elevated C-reactive protein.[3] Atypical lymphocytes may also be present in the peripheral blood.[4]
A recent PubMed search of articles published between 1972 and January
2013, revealed 734 articles that have described KFD. Keywords used
included “Kikuchi-Fujimoto disease” and “histiocytic necrotizing
lymphadenitis.” Dorfman and Berry evaluated 108 cases with a confirmed
diagnosis of KFD.[5] Of these, 88 were diagnosed in
the United States. This article provided gender, age, and ethnic
information for the total population studied but did not stratify the
information for patients in the United States alone. In addition, for
our search, those articles that did not provide the ethnicity, age, and
gender of the patients studied were not included in our review. We
focused our review on the adult population although it is known that
KFD may also occur in children.[6] A total of 22 cases with KFD have been fully described in the literature.[7-24]
In this report, we describe 3 patients diagnosed with KFD and discuss
the diagnosis, pathology, and management of this rare disease.
Case Presentation
Patient 1: Patient 1, a 26-year-old white female, presented in February 2008 with approximately four enlarged lymph nodes in the left posterior cervical area. She also complained of joint pain and fever that lasted approximately 2 weeks. Laboratory data were reportedly normal. She was initially diagnosed with infectious mononucleosis. She again presented 2 weeks later complaining of cervical lymphadenopathy and concurrent joint pain. Laboratory data at that time revealed leukopenia and thrombocytopenia. She was subsequently referred to a hematologist. She was hospitalized and underwent a comprehensive work up, with tests revealing a white blood cell count of 2.0 (103/mm3), with a total lymphocyte count of 900 cells/µL and total granulocyte count of 1000/µL, hemoglobin level of 12.9 g/dL, mean cell volume of 91.5 fL, and platelet count of 100,000/µL. HIV-1 and -2 screening tests were non-reactive, and anti-DNA and anti-nuclear antibodies were negative. Epstein-Barr Virus (EBV) and cytomegalovirus (CMV) tests were negative. Bone marrow biopsy was without evidence of malignancy and showed normal cytogenetics (46,XX). Chest X-ray suggested chronic bronchitis with minimal peribronchial thickening. CT scan of the abdomen and pelvis without contrast revealed mild hepatosplenomegaly, with the spleen measuring at least 17 cm in length. Results also suggested some dilation of the portal venous system, as well as bibasilar infiltrates with a small amount of pleural reaction. A repeat chest X-ray confirmed the interval development of bibasilar infiltrates that were worse on the left side, suggesting pneumonia. At that time, she was treated with intravenous antibiotics. Follow-up chest X-ray showed no active infiltrates. No adenopathy was noted in the chest on any study that was performed. PET/CT scan showed hypermetabolic adenopathy bilaterally in the neck, left supraclavicular, and left subclavian areas. The largest lymph node measured 2.4 cm in the anterior triangle of the left neck. The SUV maximums ranged from 3.7 to 9.8. There was no hyper-metabolism in the axillary, mesenteric, retroperitoneal, or pelvic areas. Patient 1 underwent an excisional biopsy of a left cervical lymph node. The initial differential diagnosis considered aggressive lymphoma, and she was recommended to start chemotherapy. However, she was referred for a second biopsy, and the diagnosis of KFD was suggested. Flow cytometry performed on the lymph node specimen did not identify a clonal B-cell population. The lymph node biopsy slides were reviewed at our institution. A section of the node showed extensive geographic area of cell necrosis with a rim of histiocytes and immunoblasts. Frequent single cell apoptosis was noted. Rare neutrophils were identified. Frequent plasma cells and plasmacytoid monocytes were also noted (Figure 1). These features were consistent with the diagnosis of KFD. Spontaneous resolution of the lymphadenopathy occurred within 4 months of diagnosis.
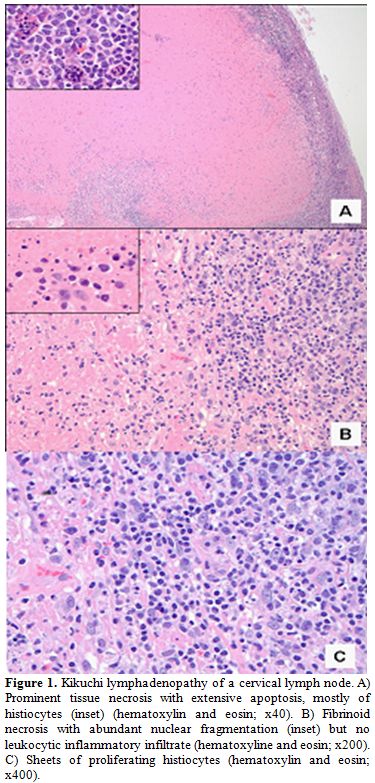 |
Figure 1. Kikuchi lymphadenopathy of a cervical lymph node. A) Prominent tissue necrosis with extensive apoptosis, mostly of histiocytes (inset) (hematoxylin and eosin; x40). B) Fibrinoid necrosis with abundant nuclear fragmentation (inset) but no leukocytic inflammatory infiltrate (hematoxyline and eosin; x200). C) Sheets of proliferating histiocytes (hematoxylin and eosin; x400). |
Patient 2:
Patient 2, a 21-year-old female, presented with a 1-year history of
generalized achiness, chronic sinus infections, chronic mouth
ulcerations, and recurrent strep infections that required treatment
with multiple antibiotics. She later developed mesogastric pain.
Physical examination demonstrated abdominal tenderness with palpation.
CT scan revealed mesenteric adenopathy. She was then referred to a
surgeon for laparoscopic biopsy of an accessible lymph node. Upon
evaluation by the surgeon, she had new onset epigastric pain, fatigue,
and night sweats that were getting progressively worse. She also had a
3-week history of diarrhea, stomach upset, cough, and pain after
urination. Labs at that time revealed white blood cell count of 16,000,
hemoglobin level of 13.1 g/dL, platelet count of 346,000/µL,
polymorphonuclears at 83%, and lymphocytes at 8.7%. Physical
examination performed by the surgeon revealed 1 submandibular lymph
node on the left side and 1 small (1 cm) cervical lymph node in the
right cervical chain. The patient underwent a laparoscopic biopsy, and
a 2 x 1.4 x 1.2 cm lymph node was removed. Intraoperatively, the lymph
nodes showed diminished cortical follicles with patent sinuses. Initial
pathological review suggested aggressive lymphoma. Upon review for
diagnostic confirmation, the histology revealed patchy areas of
necrosis consisting of brightly eosinophilic fibrinoid deposits,
including nuclear fragments and apoptotic bodies, surrounded by large
collections of pale-staining histiocytes. At the periphery of the
necrotic areas, there were nests of plasmacytoid monocytes and
immunoblasts. Neutrophils and eosinophils were absent. The findings
supported the diagnosis of KFD. The patient continued to have residual
mesenteric adenopathy for approximately 2 years following diagnosis but
this has since resolved completely.
Patient 3:
Patient 3, a 33-year-old female, initially presented with a right neck
mass approximately 1 year before evaluation. Six months later, she
developed persistent night sweats and diffuse pruritus. A fine-needle
aspiration of the right supraclavicular mass was completed, and the
final diagnosis found an atypical lymphoid hyperplasia with an
indeterminate gene rearrangement. The pathology recommended additional
biopsy to rule out lymphoproliferative disorder. An open biopsy of the
right supraclavicular lymph node was performed. The final pathology
report suggested lymphoproliferative disorder. Initial findings
included an atypical lymphoid proliferation with necrosis. The
immunohistochemistry revealed CD20 positive, CD10 positive, CD3
positive, CD43 positive, and CD30 positive in small clusters of cells
and CD15 positive in scattered neutrophils. Flow cytometry found no
clonal B-cell population by immunophenotypic criteria. The CD4/CD8
ratio was unremarkable. A PET scan found a single metabolically active
right supraclavicular lymph node. Patient 3 underwent comprehensive
staging studies, including viral studies on peripheral blood, which
were negative, and bone marrow biopsy, which was negative for
involvement of lymphoma. She subsequently underwent surgical excision
of the solitary lymph node. The findings confirmed KFD. After excision
of the lymph node, repeat CT scan was without lymphadenopathy. The
patient has exhibited no recurrent lymphadenopathy and all other
symptoms resolved.
Discussion
Physical Presentation and Diagnostic Findings of KFD: Patients may present with fevers, chills, weight loss, arthalgia, splenomegaly, and skin rash.[23]
However, the most common presenting symptom is localized
lymphadenopathy. Most commonly, lymphadenopathy is located in the
jugular carotid chain and the posterior cervical triangle, although
there may be generalized, diffuse lymphadenopathy.[25,26] Lymph nodes are usually 3 cm or less in diameter; however, they may reach 5-6 cm in diameter.[27] Additional findings may include increased lactate dehydrogenase, leukopenia, and elevated serum transaminases.[28] Approximately 25-58% of patients experience leukopenia and 2-5% of patients experience leukocytosis.[29] Elevations in erythrocyte sedimentation rate have also been noted.[26]
Excisional biopsy of a representative lymph node is the preferred way
to obtain a sample of tissue for microscopic evaluation. Skin
manifestations have been identified in approximately 30% of KFD
patients. These manifestations are non-specific and include acneiform
eruptions, facial erythema, indurated, erythemic papules and plaques,
purpura, and nodules.[31-33] Occasionally, patients will experience lip or eyelid edema and oral ulcers.[31]
Biopsy of any of the cutaneous lesions usually reveals leukoclastic
vasculitis, superficial and deep lymphohistiocytic perivascular
infiltrates with nuclear debris, papillary edema, and absence of
neutrophils.[27,31] Skin lesions are indicative of a more severe clinical presentation, such as presence of liver dysfunction.[32]
Histopathologic findings indicative of KFD include partially preserved
nodal architecture with expansion of the paracortex by patchy areas of
fibrinoid necrosis with marked apoptosis and nuclear debris, surrounded
by aggregates of histiocytes with crescentic nuclei, activated
T-lymphocytes (immunoblasts), plasmacytoid monocytes, and
characteristic absence of neutrophils and eosinophils.[25,29]
Plasmacytoid monocytes with interspersed karyorrhexis and
crescent-shaped histiocytes have been considered minimum diagnostic
criterion for KFD.[30] Histiocytic proliferation
tends to be more characteristic of KFD than necrosis alone.
Differential diagnoses that should be considered include lymphoma,
systemic lupus erythmatous (SLE), tuberculosis, and infectious
mononucleosis. Because there are no diagnostic laboratory studies
available for KFD, it is important to exclude other causes of
necrotizing lymphadenopathies, as its course and treatment are entirely
different.[29] To assist in narrowing the
differential diagnoses, the absence of auto-antibodies including anti
nuclear auto-antibodies may help to exclude autoimmune disorders.[25]
In the early proliferation stage of KFD, the presence of clusters of
large atypical cells and immunoblasts may mimic lymphoma.[31] In lymphoma, however, necrosis may or may not be extensive, and neutrophils and granulomata tend to be absent.[31] Of note, lymphoma is incorrectly diagnosed in approximately 30% of cases of KFD.[29]
Immunohistochemical staining may reveal a predominance of CD8-positive
lymphocytes.[30] These stains assist in differentiating KFD from
lymphoma.[3] In large B-cell lymphoma, neoplastic
cells are CD20 positive, whereas the large cells in KFD are
CD8-positive immunoblasts and CD68-positive histiocytes.[3,26,30]
Etiology/Pathogenesis of KFD:
The exact cause of KFD is unknown; however, it has been hypothesized
that KFD may be the result of a viral infection or a manifestation of
an autoimmune disease.[25] Infectious causes have
been suspected due to the self-limiting nature of KFD. The presence of
reactive histiocytes and tubuloreticular inclusions identified under
electron microscopy, in addition to an elevation of antibodies against
an antigen, has supported a viral infection as the source of KFD. The
tubuloreticular inclusions were also noted in endothelial cells and
lymphocytes in patients diagnosed with SLE.[31] Due
to the presence of reactive histiocytes and atypical lymphocytes, viral
infections such as Epstein-Barr virus (EBV) and human T lymphocyte
virus-1 (HTLV-1) were identified as possible causes of KFD.[34]
In situ hybridization, polymerase chain reaction (PCR), and
immunohistochemistry revealed association between HTLV-1 infection and
the development of KFD.[34] Hudnall et al.[35]
evaluated 30 lymph nodes involved by KFD. The results showed that
herpes simplex virus 1, varicella zoster virus, and human herpes virus
8 (HHV-8) DNA were not detectable; and herpes simplex virus 2,
cytomegalovirus, HHV-6, and HHV-7 were only detected on occasion.
Therefore, it is unlikely that these viruses are involved in the
pathogenesis of KFD. EBV was detected in many of the KFD samples;
however, it is also important to note that recent EBV exposure may lead
to positive serologic studies even if there is no active EBV infection.[35]
EBV titers may be elevated in reactive lymphoid tissues and are not
indicative of an active infection of EBV.[28] Elevated viral titers may
persist long after resolution of the infection. Truly relevant
serologic results include a 4-fold increase in the IgG titer or the
detection of an elevated IgM titer occurring during the disease
process.[28] Cho et al.[36] evaluated 50 lymph node specimens that were
affected with KFD for the presence of HHV-6, -7, and -8. In this study,
PCR analyses failed to establish a correlation between the presence of
HHV and KFD.[36] Zhang et al.[37]
demonstrated that parvovirus B19 may be implicated in KFD and
identified an association between parvovirus infection and KFD. A
strong correlation between SLE and KFD has been established. SLE may be
associated with fever, arthralgia, and lymphadenopathy, features that
are also characteristic of KFD.[25] Lymphadenopathy
has been reported in 23-34% of patients with SLE, and these patients
were more likely to have increased SLE disease activity levels.[25]
Lymphadenopathy associated with SLE is widespread, whereas
lymphadenopathy associated with KFD is more localized, usually only
affecting the cervical lymph nodes. Histologically, lymph nodes
evaluated in SLE and KFD are very similar and demonstrate nonspecific
changes with the exception of necrosis.[25,31]
The presence of increased vascularity, scattered immunoblasts, plasma
cells, and hematoxylin bodies are also indicative of SLE-affected
tissue.[25,31] The presence of
hematoxylin bodies is unique to SLE; however, it is rarely observed in
affected specimens. Therefore, histopathological findings are not
always definitive in establishing a diagnosis. The presence of large
numbers of plasma cells encompassing necrotic foci is also
histologically indicative of SLE.[29] Other
pathologic findings in patients with SLE may include malar rash,
proteinuria, photosensitivity, auto-antibodies, and the presence of
anti-native DNA antibodies.[26,27] There is yet to be an identifiable cause of KFD and its symptoms.
Treatment of KFD: KFD is a self-limited, benign condition that
spontaneously resolves within 1-4 months; however, 3-4% of patients
will experience recurrent episodes of KFD.[26] Due to the correlation between KFD and SLE, patients presenting with KFD should be evaluated for the presence of SLE.[25] It is also advised that, following a diagnosis of KFD, the patient should be monitored for the development of lupus.[25,33]
Treatment is unnecessary and not recommended unless there is
coincidence with SLE. For complicated cases of KFD, glucocorticoids,
alone or in combination with hydroxychloroquine, may be the treatment
of choice.[22]
Conclusion
This disease usually affects women in the age range of 20-35 years. The
female-to-male ratio is approximately 4:1. Also, there is noted to be a
higher incidence in Asiatic populations. Of the 22 adult cases
described in the United States, ten (45%) were male, and 12 (55%) were
female, with ethnicity showing 29% Asian American, 24% African
American, 19% Caucasian, 14% Hispanic, and 14% Western Asian. Age
ranged from 6 to 63 years with a mean age of 34 years. Initial
presentation was typical for most patients (lymphadenopathy, fever, and
fatigue, with most experiencing spontaneous resolution of their
symptoms within 6 months after diagnosis).[7-24] The
patients described in this case report were female with an age range of
20-35 years of age and they presented with clinical characteristics
including lymphadenopathy, pain, and sweats. The patient in case number
2 was interesting in that irritable bowel syndrome (IBS) could have
been included in her initial differential diagnosis due to the severe
abdominal pain, diarrhea, and upset stomach that she experienced.
Imaging studies revealed mesenteric adenopathy that would not be
expected with IBS. Furthermore, lymph node biopsy solidified the
diagnosis of KFD.
For patient 1, spontaneous resolution of lymphadenopathy occurred
within 4 months of diagnosis. Patient 2 continued to have residual
mesenteric adenopathy for approximately 2 years following diagnosis.
Through careful diagnosis patients with KFD may avoid unnecessary
treatment with aggressive chemotherapy and its associated toxicities,
which include the risk of secondary malignancies.
References

[TOP]