Received: January 9, 2014
Accepted: March 8, 2014
Meditterr J Hematol Infect Dis 2014, 6(1): e2014027, DOI 10.4084/MJHID.2014.027
This article is available on PDF format at:

Federica Bozzano1, Francesco Marras2 and Andrea De Maria1,3
1
University of Genova, Italy
2 Istituto G.Gaslini, Genova, Italy
3 IRCCS AOU S.Martino-IST-Genova, Italy
|
This
is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract MTB
ranks as the first worldwide pathogen latently infecting one third of
the population and the second leading cause of death from a single
infectious agent, after the human immunodeficiency virus (HIV). The
development of vigorous and apparently appropriate immune response upon
infection with M. tuberculosis in humans and experimental animals
conflict with failure to eradicate the pathogen itself and with its
ability to undergo clinical latency from which it may exit. From a
clinical standpoint, our views on MTB infection may take advantage from
updating the overall perspective, that has quite changed over the last
decade, following remarkable advances in our understanding of the
manipulation of the immune system by M. tuberculosis and of the role of
innate components of the immune response, including macrophages,
neutrophils, dendritic cells and NK cells in the initial spread of MTB
and its exit from latency. Scope of this review is to highlight the
major mechanisms of MTB escape from immune control and to provide a
supplementary translational perspective for the interpretation of
innate immune mechanisms with particular impact on clinical aspects.
|
Introduction
Mycobacterium
tuberculosis (MTB) may be regarded as the most successful
intracellular
bacterium worldwide, in view of its world prevalence and distribution.
MTB ranks as the second leading cause of death from a single infectious
agent, after the human immunodeficiency virus (HIV). Indeed, about 8.7
million incident cases of pulmonary tuberculosis (TB) (range, 8.3
million to 9 million), equivalent to 125 cases per 100,000 population,
were registered globally in 2011.[1]
Despite availability of
antituberculous drugs for the last 50 years Mth is responsible for 1.5
million deaths every year with about one third of the world population
having been in contact and latently infected.[1,2]
Since its first characterization by Robert Koch at the end of the 19th
century,[3] intense efforts have
led to the characterization of
the manifold interactions of M. tuberculosis with the immune system, to
a renewed study of its metabolism for the identification of new
specific pathways subject to inhibition by new drugs, to the discovery
of the mechanisms it uses to divert host defences and to the
understanding of the broad spectrum of soluble factors and cells
involved in its control.
Despite relevant advances over the last 20 years in our understanding
of the broad outlines of mechanisms contributing to protective immunity
to M. tuberculosis, relevant scientific and clinical challenges remain.
The development of vigorous and apparently appropriate immune response
upon infection with M. tuberculosis in humans and experimental animals
conflicts with failure to eradicate the pathogen itself and with its
ability to undergo clinical latency from which it may exit causing the
bulk of overt clinical tubercular disease in everyday clinical life. In
particular, our incomplete understanding of mechanisms potentially
allowing complete eradication of M. tuberculosis once infection has
taken place, and of those failing during latency - thus leading to
reactivation of M. tuberculosis only in a subset (10-15%) of latently
infected subjects[4] - represent
major hurdles towards effective
second-generation vaccines and targeted treatment of latency.
The classical view of immunity to M. tuberculosis mainly recognizes
participation of macrophages and cells of the adaptive immune system
(CD4+ and CD8+ T lymphocytes) in the control of mycobacteria. From a
clinical standpoint, our views on MTB infection may take advantage from
updating the overall perspective, that has quite changed over the last
decade, following remarkable advances in our understanding of the
manipulation of the immune system by M. tuberculosis and of the innate
component of the immune response. Over recent years, it has indeed
become clear, that, in addition to adaptive mechanisms, innate immune
responses are recruited by and against M. tuberculosis according to the
time-frame of response recognizing early and late events after MTB
entry.
The purpose of the present review is not to provide a comprehensive
review of TB immunology, nor to provide in depth focus on the
mechanisms of M. tuberculosis virulence and pathogenicity which appear
elsewhere[5,6] rather, scope of
this review is to highlight the
different actors of the immune response against M. tuberculosis and the
major mechanisms of MTB escape and to provide a supplementary
translational perspective for the interpretation of innate immune
mechanisms with particular impact on clinical aspects. Renewed
frameworks of interpretation of results from human and animal research
and from clinical observations will help the updating and understanding
of M. tuberculosis immunopathogenesis and facilitate the design of new
vaccines, drugs and prevention strategies.
Clinical Correlates of the Immune Response to M. tuberculosis
Active Tuberculosis (TB) encompasses a range of clinical presentations
and disease courses. Active TB occurs in two stages, either as the
natural evolution of overwhelming M.
tuberculosis replication following
initial infection (Primary or primary-progressive TB), or resuming
after a latent infection/containment of M. tuberculosis
that may last
many years following exposure (post-primary TB or reactivated TB). Both
primary and post-primary TB occurs in only a minor fraction of those at
risk, as a consequence of several factors that include both innate and
adaptive immune responses. Primary TB is detected in up to 20% of those
exposed to M. tuberculosis airborne inoculum, and post-primary TB with
reactivation of M. tuberculosis from latency occurs at a rate of
0.1-0.5% per year with an estimated 5-10% lifetime risk of developing
active TB.[4,7]
This heterogeneity of individual responses is
associated to different immunogenotypic characteristics (extending from
innate immune responses to adaptive immune control of M. tuberculosis)
and is highlighted by the non-human primate model (Cynomolgus macaque,
Macaca fascicularis)
of experimental bronchial inoculation of a fixed
MTB inoculum.[8] Here, a whole
spectrum of outcomes and pathological
findings were observed, similar to what occurs after acute infection in
humans. The outcomes of instillation of a defined inoculum was
invariably infection, however the spectrum of pathology included
macaques that progressed rapidly and succumbed to active disease,
others that developed active disease over a more chronic course
(including one who spontaneously resolved the infection), and those
that displayed no evidence of disease even though they were clearly
infected and had clinical characteristics similar to latent TB in
humans. The heterogeneity of the host immune response extends beyond
primary TB, and applies particularly to latent TB in humans -where only
between 20-50% of latent close contacts of TB cases develop Tubercolin
Skin Test (TST) reactions and 1-2% of these close contacts eventually
develop active TB[9,10] - and to
latent TB in cynomolgus
macaques.[8,11]
Thus, latent TB is reflective of a heterogeneous group
of individuals:[12] a) those who
have subclinical disease,
b) those who
will progress to primary
active disease; c) those who maintain persistent, lifelong infection;
d) those who temporarily suppress
infection but later develop active TB, possibly as a result of
immunosuppression or some other event (i.e., true latent infection);
e)
those who are able - either through innate or adaptive immunity or the
combination - to effectively clear the pathogen (Figure 1).
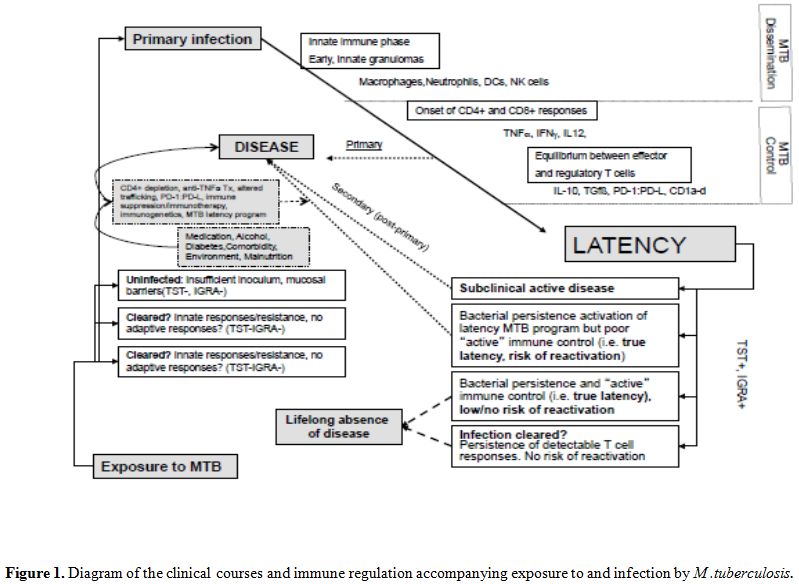 | Figure 1. Diagram of the clinical courses and immune regulation accompanying exposure to and infection by M. tuberculosis. |
Unfortunately, no
test is currently available to differentiate latent
from active TB disease, as TST and interferon gamma-release assays
(IGRA) simply report the presence of specific T cell responses
irrespective of the clinical condition. Furthermore, there is no test
to identify those latently infected individuals who may progress to
active TB or those who have subclinical disease. Also in this case TST
and IGRA do not help discriminate different clinical courses.
The ability to identify those individuals with latent TB who are at
risk of reactivation would help target preventative therapy and devise
individualized target treatment thereby increasing adherence and
minimizing toxicity and costs. Using transcriptional profiling of
leukocytes in whole peripheral blood by microarray analysis, a
characteristic neutrophil-driven IFN-inducible 393 transcript-signature
has been identified in patients with active TB.[13]
This transcript
signature disappears by 2 months of effective treatment and correlates
with the extent of radiographic involvement. Interestingly, 10-20% of
patients with TB latency have a transcript signature similar to those
with active disease. Although not proven yet, these patients might
represent the minority of latent TB who will eventually progress to
active TB years later. Leukocyte or purified cell population
transcriptional analysis - also in other areas of chronic infections
including HCV[14] - is likely to
become a useful future tool to
identify the subset of patients with true latency who will develop
post-primary reactivation and for whom chemoprophylaxis - or rather
treatment - may be mandatory. The precise labelling of patients with
different types of latency using molecular or immunological tools
represents one of the main future challenges to individualize treatment
of smoldering infection, prophylaxis of true latency and avoiding
unnecessary toxicity for eradicated infection.
Timing of Immune Responses and Granuloma Formation
Following the establishment of M. tuberculosis infection in the airways and lung parenchyma, the bacilli are believed to be phagocytosed by the alveolar macrophages[15] and are taken up by neutrophils[16] and dendritic cells (DCs).[17] Over time, cells progressively assemble in a compact, organized aggregate of mature macrophages surrounded by fibroblasts and interspersed with neutrophils, DCs, Natural Killer cells, B cells, CD4+ and CD8+ T cells. This structure is a granuloma and has been historically and until recently considered to represent a concentrated effort of the immune system to sequester, wall off and eradicate M. tuberculosis.[18,19] Recent evidences have however subverted the classic view that the granuloma is a host-protective structure. Indeed, different stages of the immune response to M. tuberculosis can be recognized, and granulomas are dynamic structures that are initially exploited by the bacterium to subvert the immune response, replicate and spread at other locations.
Innate immune phase-granuloma dynamics. Upon MTB entry in the airways innate immune responses predominate. Early granulomas are composed of inflammatory macrophages, neutrophils and DCs that progressively accumulate upon recruitment. All the cells of the early granulomas engulf the mycobacteria and become infected. Pathogenic mycobacteria such as M. tuberculosis and M. marinum in the zebrafish model have evolved multiple mechanisms to manipulate this cellular niche to their own advantage. Trafficking and maturation of phagosomes in which pathogenic mycobacteria reside is manipulated to prevent lysosomal killing and degradation.[20] Surprisingly, in spite of overwhelming infection, macrophages and DCs in the early granuloma are inefficient in presenting M. tuberculosis antigens in the early granuloma to CD4+ T-cells.[21] Efficient MTB Antigen (Ag)-presentation only takes place later in the lymphnode.[22] Mycobacteria, as exemplified by the zebrafish-M. marinum model, exploit granuloma formation for their proliferation and dissemination in the infected host. Dynamic imaging studies reveal that macrophage move rapidly within granulomas, at speeds comparable to lymphocytes in a chemokine gradient.[23] Movement is dictated by the RD1 virulence locus that is responsible for M. tuberculosis ESX-1 secretion system (which is lost in attenuated Bacillus Calmette-Guerin),[24,25] and ceases when macrophages contact dying infected cells, thereby increasing the number of infected cells. Also the necrotic core of granuloma, which was regarded as being not involved in immune interactions, is crossed by infected and non-infected macrophages.[21,26] Finally, tertiary lymphoid structures are found in granulomas in the lungs of mice.[27] Here, macrophage and T-cell movement resemble those of T and B cell trafficking in secondary lymphoid organs,[21,28] and chemokines are produced (CCL19, CCL21), which are characteristic chemoattractants for CCR7-bearing lymphocytes homing to lymphoid structures.[29] Thus, despite macrophage, T-cell and DC entry into the granuloma in the early phase, these cells do not leave, and at the same time cannot proceed to antigen presentation neither locally nor in lymphnodes.
TNF-alpha vs. MMP-9 in granuloma formation. During early granuloma formation, TNFα has been historically considered instrumental to granuloma formation and to increase the ability of macrophage control of intracellular mycobacteria.[30] This view is however challenged by the observation that in non-human primates TNFα-blockade results in disseminated disease with normal granuloma structure,[31] and by similar findings in patients treated with anti-TNFα treatment.[32,33] Indeed in the zebrafish model, TNFα increases pathogenic M. marinum death and its absence is associated with accelerated and increases granuloma formation.[34] Rather than TNFα, the mechanism(s) underlying granuloma formation have been shown to involve induction of host matrix-metalloprotase-9 (MMP-9) production by macrophages and epithelial cells upon interaction with RD-1 locus-encoded, secreted ESAT-6.[25,35] In line with this view, MMP-9 knockout mice have decreased granuloma formation and improved control of infection and has been found to be enriched in tissues and pleural fluid in human pulmonary TB.[36]
Death
matters: Apoptosis vs. Necrosis.
During early phases, mycobacterial load is rapidly rising through
granuloma formation with influx and infection of neutrophils and
macrophages and cell death. While necrosis, with cell lysis, propagates
locally viable mycobacteria and increases pathogen load, programmed
cell-death or apoptosis maintains intact cellular membranes favoring
cellular compartmentalization and mycobacterial containment.[37,38] The
type of cell death that is induced depends on the regulation of the
lipid mediator eicosanoids prostaglandin E2 (PGE2, proapoptotic) and
lipoxin A4 (LXA4, pronecrotic).[39]
Differences in eicosanoid pathway
activity and regulation may contribute to inter individual differences
in cross-presentation of M.
tuberculosis by Dendritic Cells (DC), thus
affecting also adaptive immune differences and the clinical evolution
from infection to primary disease or to true latency.[40]
Neutrophils
support M. tuberculosis
replication and spread[16,41] and may have dual
roles in the early defense against the pathogen. Activation of
antigen-specific CD4+ T cells is facilitated by neutrophils,[42]
however inhibition of neutrophil apoptosis by MTB determines their
delayed activation.[43]
Therefore early - and sometimes late - granuloma formation does not
“wall off” mycobacteria. The view of a mechanical containment in
granulomas following TNFα induction is being replaced with a new
perspective indicating that granuloma formation is induced by
pathogenic mycobacteria through mechanism(s) including
ESAT-6-induced[25] MMP-9
production.[35] Thus early
granulomas favor
increased macrophage accumulation, mycobacterial replication, and
systemic MTB spread. Even adoptive transfer of Ag-specific CD4+ T cells
shows that in this phase of the infection Mycobacteria are secluded in
a protected niche within the granuloma[44]
and that intervention should
target innate immune events (including anti-MMP-9[45]
or pro-apoptotic
treatments), that predominate during the early phase but that persist
also in later equilibrium phases of the infection.
Overall, therefore, the early stages of antimycobacterial immune
responses are dominated by innate immune responses that have little
immediate antimycobacterial effect and rather favor its spread and
replication. The subsequent adaptive phase however builds on initial
innate responses, which are eventually needed for antigen presentation
and editing of adaptive responses.
Adaptive Responses and Immune Equilibrium
The relatively small proportion of patients that progress to primary TB
following infection by M.
tuberculosis and of those that upon acquiring
latent infection progress to post-primary disease should be regarded as
a success of host defenses, even if latency consists in arrest of
bacterial growth, not in bacterial sterilization.
The prominent characteristic of specific antimycobacterial adaptive
responses is the long delay in onset and the need for their continuous
persistence and effort to maintain latency. Adaptive responses are
relevant to containment and control of MTB replication, involve
IFN-γ-producing or poly-functional (IL-2, IFN-γ and TNFα) CD4+ and CD8+
T lymphocytes. Adaptive responses are delayed in the early
granuloma, and ultimately rely on the presentation of specific
mycobacterial antigens by DCs, under editing, control and help by NKT
and NK cells.[46,47] Initiation of
the adaptive response begins in
lymphnodes, where infected DC traffic after initial delay and
persistence in peripheral tissues (alveoli and lung tissue) where even
100-fold higher bacterial concentrations are found.[22,48] Further
local delay in lung adaptive responses to M. tuberculosis is
due to the
influx of pathogen-specific CD4+ regulatory T cells generated in
lymphnodes ad migrating to the tissue,[49]
and by the direct inhibition
of apoptosis by M. tuberculosis in infected neutrophils.[43] Regulatory
CD4+ T cells (Treg) are generated in lymphnodes together with Th1 (T
helper 1) CD4+ T cells in the early phase of adaptive responses, and
are responsible for failure to eradicate M. tuberculosis in the long
run, as shown by adoptive transfer in the mice model.[50]
In addition
to the presence of Treg CD4+ T cells, also the expression of PD-1 on
Ag-specific CD4+ T cells is a factor favoring M. tuberculosis
persistence and survival once latency has been established in the mice
model.[51] Overall, however,
survival to M.
tuberculosis relies on the
presence of CD4+ T cells which play a fundamental role in inhibiting
its replication and protect from active disease. Indeed CD4 lymphopenic
patients with or without HIV infection are at increased risk of
developing active TB.[52] Although
CD4+ T lymphocytes have been
considered to be the primary source of IFN-γ and to be protective
through its secretion, this is not the case. CD4+ T cell depletion
induces disease in mice while leaving unchanged lung tissue levels of
IFN-γ.[53] and conversely, IFN-γ
deficiency still allows
protection.[54] In mice and human
models it appears that CD4+ T cells
per se, rather than their production of IFN-γ may be protective. High
IFN-γ levels in lung tissue and granuloma may be attributed to
Ag-specific CD8+ T cells which produce IFN-γ and TNF-α and are involved
in the control of M.tuberculosis,[55] and to NK cells, that are the
main IFN-γ producers involved in DC maturation and editing.[56]
HLA-E-dependent presentation of peptides to human CD8+ T cells may be
also involved in the control of M.
tuberculosis and has been found to
comprise a dominant immune response in latently infected patients.[57]
Additional support for CD8+ T cell involvement in the control of M.
tuberculosis is provided by the mycobacteriostatic effect
of granulysin
in CD8+ CTL granules[58,59] and by
disseminated infection in mice
lacking HLA class I presentation pathways (e.g. ß2-microglobulin,
transporter associated with antigen processing, TAP).[60-62]
TB Reactivation
One of the most obscure areas in our understanding of the relationship
of the immune system with M.
tuberculosis is represented by the
clinical transition from latency to post-primary TB. Factors underlying
this transition are so far only partially understood, and there is no
understanding of the precise mechanism(s) that induce transition to
reactivation from a dormant state. Knowledge of these mechanism(s)
would be of crucial importance, since this would allow i)
identification and prophylactic/therapeutic targeting of the minority
of patients with latent TB that will actually progress to reactivation,
thus avoiding unnecessary potentially toxic and costly courses of
chemoprophylaxis administered to those who would never need it in a
lifetime; ii) monitoring and prediction of the exact moment when M.
tuberculosis would exit latency in these patients and iii)
devise
immune intervention/vaccination strategies to boost immune control of
M. tuberculosis of this selected minority of patients.
Although it has been held for a long time that the merit for latency
persistance should be attributed to the immune system, evidences
accumulated in the last years point out that also M. tuberculosis
actively participates to this process. M. tuberculosis
activates a
bacterial regulon controlled by the DosR-DosS signal transduction
system in the presence of local hypoxia, carbon monoxide or nitric
oxide.[63,64] In the presence of
these stimuli, which are believed to
be prevalent during latency, M.
tuberculosis activates the expression
of a set of genes allowing the use of alternative energy sources.
Within this program, it expresses genes whose products are recognized
by T cells. The regulation of this set of genes during latency and the
shut off of the transcriptional program that is active during the
replicative active phase of M.
tuberculosis life cycle implies that
specific mycobacterial epitopes become available during latency while
others may disappear and no longer be recognized.[65,66]
M. tuberculosis
encodes two additional gene clusters that are involved
in exit from latency, whose regulation contributes to determine the
outcome of infection. Five encoded proteins resemble the
“resuscitation-promoting factor (Rpf)” produced by M. luteus to
recover
from nutrient-starved latent phase.[67]
Deletion of Rpf-like genes of M.
tuberculosis impairs mycobacterium recovery from latency[68,69] in a
mice model. Interestingly, double RpfAB knockouts have a different
interaction with innate immune mechanisms and induce higher amounts of
TNFα and IL-6 in infected macrophages. Rpf-like gene products therefore
provide a clockwork for exit from latency and also a system to modulate
innate responses thus favouring mycobacterial growth. Finally, an 88
toxin-antitoxin gene pair system is also encoded by M. tuberculosis and
its transcription is involved in the decision to maintain latency or
progress to overt replication and virulence.[70]
In view of the above data, shedding further light on the fine-tuning
mechanisms employed by M. tuberculosis to regulate its access to and
exit from latency represents a crucial step with relevant clinical and
immune bearing. Immunoprophylactic prevention of exit from latency may,
for example, require different antigen and epitope-targeting compared
to those encoded by pathogenic replicating bacteria during primary
invasion. Also, targeting of some of these transcripts/proteins may
provide new tools for antibacterial treatment of early reactivation.
In general, exit from latency into post-primary TB is regarded as a so
far poorly characterized consequence of “immune weakening”, and
represents and event that is not predictable according the when, who,
and where questions. As mentioned above, among any cohort of latently
infected subjects it is impossible to predict who will fall in the 10%
that eventually will experience reactivation, when this will take place
or where the escape from immune control and exit from M. tuberculosis
latency program will eventually occur (although this will occur in the
lungs in 85% of the cases due to the high mycobacterial burden in this
site).
Regardless of bacterial virulence factors involved in latency exit, two
specific well characterized mechanisms are known to increase the
likelihood of reactivation. The first involves quantitative and
qualitative depletion of CD4+ T cells, while the other is represented
by impairment of TNFα signaling. Immune deficiencies leading to CD4+ T
cell loss, including HIV-1 infection, are associated to increased risk
of M. tuberculosis
reactivation.[71] The risk of TB
reactivation during
HIV is associated not only to quantitative defects of CD4+ T-cell
counts, since many patients develop TB and AIDS well before CD4+ T-cell
counts decrease below 350-200/µl. Selective targeting of TB-specific
CD4+ T-cells, functional derangement of CD4+ T cells with skewing of
polyfunctionality (IFN-γ,TNFα and IL-2 production) or skewing of
cytokine production patterns have been advocated.[72,73,74-77]
No
precise CD4-associated mechanism has been so far pinpointed, however,
to explain how CD4+ T cells accomplish control of M. tuberculosis in
some patients but fail in others. With regard to TNFα, it has been well
established that neutralization of TNFα particularly in the context of
monoclonal antibody treatment,[78,79]
dramatically increases the
chances of TB reactivation. In vitro, TNFα production and signaling in
monocytes controls M.
tuberculosis replication,[34]
as also shown with
comparative use of RpfAB knockouts and wildtype strains in vitro.[68]
Additional mechanisms have been suggested to be involved in the exit
from latency, such as programmed death receptors and ligands
(PD-1/PD-L1). Increased blood expression of PD-L1 occurs during active
TB and is predominantly due to its expression by neutrophils.[80] In
addition, PD-1 expression is associated with different functional
profiles in CD8+ antigen specific CTLs in active and latent TB,[81]
thus suggesting different functional predominance in the two
conditions, and the crosstalk between innate and adaptive elements of
the immune response. The ability to modulate gene expression and to
shift antigenic expression during overt replication and latency may
represent one of the mechanisms of evasion from the control by the host
immune responses. The expression of different gene products during
specific and different phases of the disease course may represent a
mechanism for mycobacterial evasion from CD4- and CD8-specific T cell
responses. For example, ESAT-6 and Ag85B represent major antigenic
targets for CD4+ and CD8+ T cells and are actively expressed during
overt infection. However, M.
tuberculosis downregulates their
expression as soon as specific CD4+ T cells appear in the mouse model,
thus favoring the persistence of the pathogen.[82,83]
Altogether the increased frequency of TB reactivation in CD4 depleted
patients (e.g. HIV-infected) and in those who undergo TNFα-blocking
treatments provide evidence that these represent two major elements
contributing to persistent and successful control of M. tuberculosis replication
and should be regarded as a correlate of efficient pathogen
control. Since they represent respectively adaptive and innate arms of
host anti-infective defenses, it is tempting to consider that exit from
latency into overt disease may be due to modulation of either arm, and
possibly through as yet poorly acknowledged pathways.
Involvement of NK Cells in Early and Late Events Affecting Individual Disease Course
The attention on early innate events leading to
permissive
granuloma formation, and the subsequent development of a highly
effective control of M.
tuberculosis has so far concentrated on crucial
events in monocytes, macrophages, neutrophils and dendritic cells. NK
cell involvement has been left out of focus despite accumulating
evidences of their involvement in the path of innate mechanisms leading
to CD8+ and CD4+ adaptive responses. Several lines of evidence point to
NK cell involvement at several steps along the path of control - or
lack thereof - of M.
tuberculosis replication and spread during primary
seeding, in the control/exit from latency, and contributing to the
immune response to vaccination and to innate resistance to infection.
This paragraph is aimed at putting these aspects in frame.
Not only CD4+ and CD8+ T cells, but also NK cells have been shown to
play a crucial role in killing of M. tuberculosis in human monocytes
through production of IFN-γ.[84]
In the RAG(-/-)T-cell deficient mouse
model M. tuberculosis stimulates
NK-cell dependent IFN-γ production in
naive spleen and lung cells, and NK cell-knockout or anti-IFN-γ-treated
animals display dramatically increased susceptibility.[85]
NK cell may interact specifically with both infected macrophages and
directly with mycobacteria through multiple receptors, thus
anticipating a direct NK cell involvement in the recognition of
mycobacteria upon entry in the lung tissue and throughout later events
contributing to the generation of adaptive responses. The most
physiologically crucial direct interplay of mycobacteria with NK cells
is represented by the interaction with TLR2 (Toll-Like Receptor 2)[86]
possibly via binding to peptidoglycan.[87]
A direct contact of
mycobacterial mycolic acids and arabinogalactan with NK cell-triggering
natural cytotoxicity receptor NKp44 has also been suggested.[87,88]
More importantly, mycobacteria-infected macrophages are directly
recognized and lysed by NK cells via NKG2D and NKp46 that recognize
ULBP1 and vimentin whose expression on macrophages is upregulated upon
infection.[89-91] BCG-exposed
macrophages (M0 and M2) in addition
induce strong activation of resting NK cells in vitro leading to their
production of IFN-γ and to cytotoxic activity induction.[92] Overall,
IFN-γ is produced not only by Ag-specific CD4+ T cells during late
events accompanying establishment of adaptive immune responses, but
also by NK cells –together with TNF-α - throughout early and later
events after mycobacterial entry and spread[93,94]
and also maturing NK
cells may contribute to BCG-induced immune responses with IFN-γ
production.[95]
NK cells positively modulate adaptive immune responses against
mycobacteria, and thus contribute to the mechanisms that ultimately
lead to control of M.
tuberculosis replication through influx of
antigen-specific CD4+ and CD8+ T cells in areas of M. tuberculosis46,56]
In addition, NK cells interacting through CD40L with
Ag-specific CD8+ CTL also contribute to CD8+ CTL killing of infected
macrophages and to their IFN-γ production[96]
and to direct control of M.
tuberculosis growth.[97]
Finally, lysis of FoxP3+CD4+ Treg by NK
cells[98] provides an additional
role played by these cells in the
control of mycobacterial replication and spread thus dynamically
counteracting the influx of CD4+ Treg cells in the granuloma that
prevent early clearance of M.
tuberculosis.[50]
Negative regulatory mechanisms for NK cell function with regard to M.
tuberculosis infection have been described by the
possibility to
express de novo PD-1 and PD-L, thus dampening their activity.[99] In
addition, induction of CD1d on mycobacteria-infected monocytes
negatively modulates NK cell triggering and induction of monocyte
apoptosis and M.
tuberculosis killing through interaction with
inhibitory receptors expressed on NK cells.[100,101]
Finally, escape
from NK cell-induced killing/apoptosis and of mycobacterial killing may
occur in M2 monocytes.[92]
The spectrum of different mechanisms that may cause NK cell
intervention during M.
tuberculosis invasion and possibly during
latency, and the array of mechanisms that negatively balance NK cell
anti-mycobacterial function suggest that interindividual differences in
the regulation of NK cells may contribute, in addition to other innate
and adaptive variability, to the wide differences in clinical courses
commonly observed after acute infection and in exit from latency in
humans and in non-human primate models.[8]
In addition to be recruited
to and detected in lung tissue granulomas of TB patients, NK cells show
dramatic interindividual differences in IFNγ and TNFα production in
healthy donors with up to 1000-fold variation upon BCG or H37Rv
challenge.[94] Thus, modulation of
NK cell function, either inherent or
acquired through exogenous factors, may underlie differences in
clinical courses and thus help explain and possibly predict the disease
course. While the majority of latently infected individuals will never
experience reactivation, a fraction of patients develops re-activation
through as yet poorly understood mechanisms.[12]
In this regard,
regulation of NK cell function and receptor expression modulation could
contribute to determine exit from latency. Indeed, patients with TB at
reactivation have dramatically decreased expression of NKp30 and NKp46
natural cytotoxicity receptors.[102]
These receptors are involved
respectively in DC recognition/DC editing with downstream shaping of
adaptive responses and in the recognition of infected
macrophages.[46,47,90,92,103]
Accordingly, decreased NCR (NKp46, NKp30)
expression at reactivation is accompanied by relevant decreases in NK
cell cytotoxicity and defective IFNγ production upon activation in
vitro.[102] With specific
treatment, TB patients fully recovering
clinically display a transcriptional signature of improvement in their
PBMC,[13] recover NK cell IFNγ
production, but do not recover NKp30 and
NKp46 expression.[102]
Specific functional skewing of other cells or molecules of the innate
immune system are likely to play significant roles in this context. The
recent finding that TLR-2 and TLR-9 polymorphisms are associated to an
increased risk of TB in different populations[104,105]
possibly due to
attenuation of receptor signalling[89,106] coupled with the expression
and relevance of these two TLRs also for NK cell activation[86,107,108]
suggests that multiple non-mutually exclusive mechanisms may contribute
the events that lead to exit from latency in infected individuals.
Therefore, both acquired environmental factors as well as inherent
HLA-unrelated (e.g.: triggering receptor expression-NCR modulation) and
HLA-related (e.g. inhibitory receptor- KIR-carriage/expression)
mechanisms are likely to influence NK cell function and their
contribution to the decreased innate immune surveillance during exit
from latency.
Conclusion
An increasingly focused picture of the early events leading to disease
progression or establishment of latency has been provided in recent
years by the investigation of innate immune mechanism(s) involving
macrophages, neutrophils, DCs and NK cells, by the development of
advanced animal models and by translational research in human disease
addressing key questions on immune correlates of M. tuberculosis
infection.
All the components of innate immune responses provide relevant
contributions to the control of M.
tuberculosis by antigen-specific
adaptive T cell responses. The knowledge of these mechanism(s) will
allow future development of vaccination strategies, monitoring of
vaccine efficacy in selected individuals with specific innate immune
response patterns, and in the identification of the minority of
latently infected individuals who will develop reactivation
post-primary disease.
References

[TOP]