Received: May 2, 2014
Accepted: June 11, 2014
Meditterr J Hematol Infect Dis 2014, 6(1): e2014051, DOI 10.4084/MJHID.2014.051
This article is available on PDF format at:

Abhishek Purohit1, Mukul Aggarwal1, Roshan B Colah2, Anita H Nadkarni2 and Hara P Pati1
1
Department of Hematology, All India Institute of Medical Sciences, New
Delhi, India.
2 Institute of Immunohaematology,
Haematogenetics. Mumbai, Maharashtra, India.
|
This
is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract Hb
Fontainebleau is a rare alpha chain variant in the Indian population
which generates an unknown peak on hemoglobin HPLC study and does cause
diagnostic difficulty to those who are not acquainted with this entity.
We present a case of Hb Fontainebleau, an eighteen year old patient who
presented with symptoms related to anemia to our department and unknown
peak observed in HPLC plots lead us to family study and molecular
characterization for this case.
|
Introduction
Alpha-thalassaemia,
one of the most common human genetic abnormalities known, is inherited
as an autosomal recessive disorder. They have clinical phenotype
varying from almost asymptomatic to a lethal haemolytic anemia and can
occur due to deletional or non-deletional mutations. Non-deletional
alpha thalassemias are relatively rare, and cause more severe anemia
clinically. Hb Fontainebleau is one of the rarely reported clinically
asymptomatic α chain variants.[1]
It shows a
characteristic retention time on hemoglobin HPLC (High-performance
liquid chromatography) which may cause diagnostic dilemma if one is
unaware of its pattern. We are reporting a case of Hb Fontainebleau,
who attended our hematology clinic for symptoms related to anemia. This
patient represents the sixth case of this alpha chain variant reported
worldwide.
Case Presentation
An eighteen year old Punjabi male born to a non-consanguineous marriage
presented to hematology department with two year history of weakness
and easy fatigability. His past and family history was not significant.
Physical examination revealed pallor only. There was no icterus,
lymphadenopathy or hepatosplenomegaly. Systemic examination did not
reveal any abnormality.
Laboratory investigation revealed hemoglobin 7.2 g/dL, total leucocyte
count 6.01 x 109/L
with normal differential count and platelet count 141 x 109/L.
Peripheral smear examination revealed microcytic hypochromic red cells
with mean corpuscular volume (MCV) 68.9 fL, mean corpuscular hemoglobin
(MCH) 15.8 pg and mean corpuscular hemoglobin concentration (MCHC)
being 22.9 g/dL and red cell count 4.56 x 1012/L.
His serum ferritin was 10 ng/ml; serum iron was 12 μg/dL, total iron
binding capacity was 589 μg/dL, unbound iron binding capacity was 587
μg/dL and transferrin saturation was 2.03%. HPLC analysis (BioRad, Beta
thalassaemia short program) on the patient’s blood revealed 13.8% of an
unknown hemoglobin with retention time of 2.92 minutes which appeared
as a hump in the peak adjoining Hb A (Figure 1).
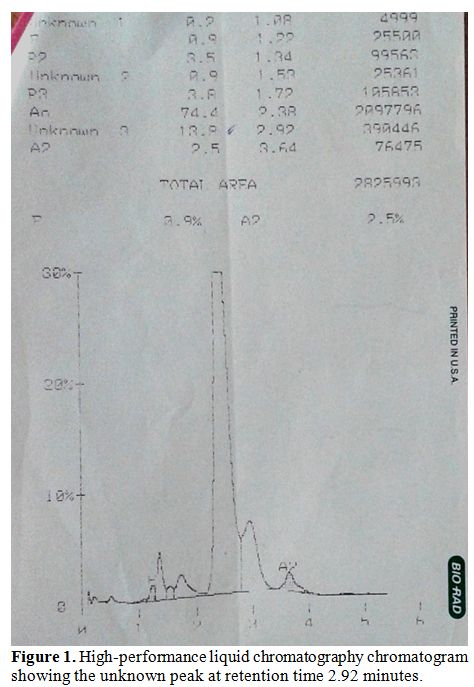 | Figure 1. High-performance liquid chromatography chromatogram showing the unknown peak at retention time 2.92 minutes |
The
other hemoglobins on HPLC were 77.6% of Hb A, 2.3% of Hb A2 and 1.2% of
Hb F. Cellulose acetate electrophoresis (pH 8.9) did not show any
abnormal band. HPLC of the parental samples was performed which
revealed the presence of a similar 11.6% unknown hemoglobin with
retention time 2.87 minutes in the mother. Her hemoglobin was 10.3 g/dL
with MCV 81.7 fL, MCH 25.1 pg and MCHC 307 g/L red cell count 4.10 x 1012/L. Father
had hemoglobin of 12.0 g/dL with MCV 59.0 fL, MCH 18.6 pg and MCHC 315
g/L red cell count 6.46 x 1012/L.
His HPLC analysis revealed raised Hb A2 (5.4%) suggesting heterozygous
beta thalassaemia.
Molecular characterization was done by automated DNA sequencing on the
ABI Prism 310 DNA sequencer (Applied Systems, Foster City, CA, USA.)
showing presence of a heterozygous G>C substitution at codon 21
(alpha 2 globin gene) leading to the substitution of alanine to proline
at the beginning of the beta helix in the alpha chain corresponding to
Hb Fontainebleau in the index case, his mother and two sisters (Figure 2).
The father was heterozygous for beta thalassaemia with the CD 8/9(+G)
mutation. The patient was treated with oral iron supplements due to low
ferritin levels and responded well to therapy with relief of symptoms
and increase in hemoglobin on the subsequent evaluation.
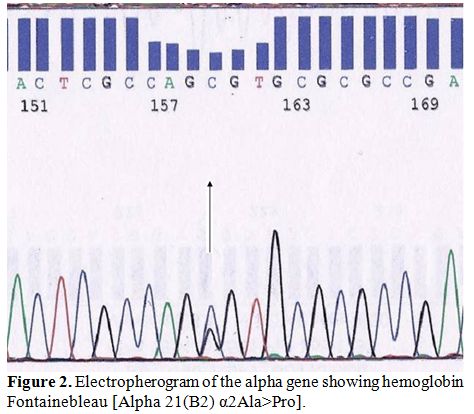 | Figure 2. Electropherogram of the alpha gene showing hemoglobin Fontainebleau [Alpha 21(B2) α2Ala>Pro]. |
Discussion
Mutations that cause alpha thalassemia can be deletional or
non-deletional types. Non-deletional alpha thalassemias are relatively
rare, result in more severe anaemia and are because of a variety of
mutations including punctual substitutions and small insertions ⁄
deletions.[2] The majority of these
mutations are
found to involve the upstream α2 globin gene proximal to the
erythroid-specific regulatory region called multispecies conserved
sequences (MCS).[3]
Many mutations have been described affecting mRNA processing, mRNA
translation, and α-globin stability. Hb Fontainebleau [alpha 21 (B2)
Ala> Pro, HBA2: c 64G>C] is an alpha chain variant
characterized
by an alanine→proline substitution at codon 21 with a GCT>CCT
change
at the DNA level, this proline residue is located at the beginning of
the beta helix.[4] Literature
search reveals only five reported cases of Hb Fontainebleau worldwide,
including two cases from India.[5-8]
The first reported case of Hb Fontainebleau was that of an adolescent
female of Italian origin, who had severe anemia. However, this severity
was explained by the co-existing membrane defect, spherocytosis with Hb
Fontainebleau.
This hemoglobin variant has electrophoretic properties identical to
those of Hb A with the exception of isoelectrofocusing in which it
migrates like Hb A1c. The introduction of a prolyl residue at the
beginning of the B helix in the alpha chain does not lead to a change
in the stability or oxygen binding properties of the hemoglobin
molecule.[4]
With identification of a second case it was observed that although
slightly unstable, this variant was expressed at 28-29% of the total
and was caused by a heterozygous mutation in the alpha2 gene. This
second reported case was an adult Iraqi male, living in New Zealand.[5] The third case was seen during
thalassemia screening in the Greek Cypriot population in Cyprus.[6]
Upadhye et al., in their newborn screening for sickle cell disorders,
identified a baby with Hb Fontainebleau in a compound heterozygous
state with Hb S and the baby had anemia at birth (Hb 11.4 g/dL),
however had no cyanosis, icterus or need for transfusion.[7]
The second case from India was a 35 year old lady, a case of secondary
infertility who presented for routine antenatal screening program for
thalassemia and had normal hematological indices.[8]
The present case was different from other reported cases from India as
it was not part of a screening programme, but rather presented due to
his moderately severe anemia. However causal relationship of anemia
with Hb Fontainebleau cannot be established as he responded well to
iron supplements and achieved near normal hemoglobin level. Further,
his mother and both the sisters were asymptomatic.
Conclusions
Hb Fontainebleau is a rare alpha chain variant in the Indian population. It generates an unknown peak on HPLC and can cause diagnostic difficulty for those who are not acquainted with this entity. However this produces no symptoms of anemia so a patient presenting with anemia and Hb Fontainebleau should be investigated for other causes of anemia and counseled properly.
References

[TOP]