Received: June10, 2014
Accepted: July 27, 2014
Meditterr J Hematol Infect Dis 2014, 6(1): e2014056, DOI 10.4084/MJHID.2014.056
This article is available on PDF format at:

Mazyar Shadman and H. Joachim Deeg
Fred Hutchinson Cancer Research
Center and the University of Washington School of Medicine, Seattle,
WA, USA, 98109-1024.
|
This
is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract The
incidence of myeloid malignancies, including myelodysplastic syndromes
(MDS) increases with age. While several therapeutic modalities have
been developed, for most of these patients the only treatment with
curative potential is allogeneic hematopoietic cell transplantation
(HCT). The development of reduced/low intensity transplant conditioning
regimens allows to successfully transplant patients in their ‘60s and
even ‘70s, although comorbidities may determine who does come to
transplantation and who does not. Also, as many as half of the patients
will develop graft versus host disease (GVHD), even with HLA matched
donors, requiring therapy for extended periods of time, and GVHD and
treatment with glucocorticoids is likely to impact the quality of life.
Nevertheless, dependent upon disease stage at HCT, the presence of
comorbidities and the regimen used, 30% to 50% of patients 60 years of
age or older, may survive long-term cured of their disease. Future
studies should focus on the incorporation of non-transplant modalities
into the overall transplant approach, the prevention of GVHD, and the
utilization of immunotherapy to reduce the incidence of relapse and
GVHD and further improve overall transplant success.
|
Introduction
The
past decade has seen explosive developments of our understanding of the
genetics and molecular biology of myeloid disorders, and new
classification schemes incorporating those insights are emerging. The
development of drugs exploiting the improved understanding of disease
mechanisms is changing therapeutic strategies and altering the natural
disease course. While those strategies for myelodysplastic syndrome
(MDS) or myeloproliferative neoplasms (MPN) are not curative, they
impact the decision regarding allogeneic hematopoietic cell
transplantation (HCT), particularly in older individuals.
Nevertheless, allogeneic HCT is currently the only modality with proven
curative potential. As increasing age is associated with a greater
prevalence of comorbidities, HCT had been reserved, until recently, for
younger individuals. The development of novel strategies now allows to
transplant, successfully, patients in the seventh or even eighth decade
of life. However, HCT is not without potential problems including
post-transplant relapse, and graft-vs.-host disease (GVHD). These
issues raise ethical questions, particularly in regards to quality of
life (QOL) vs. quantity,
and with the availability of non-transplant
treatment options that modify the natural disease course, patients (or
physicians) may prefer treatment, for example, with hypomethylating
agents, rather than proceeding to HCT. There are also socioeconomic
issues and questions of health resource utilization. Both transplant
and non-transplant modalities are cost-intensive, and treatment cost
and insurance coverage are issues that need to be addressed. We will
review here some aspects of HCT in older individuals with a focus on
MDS.
Background
The spectrum of MDS ranges from very low-grade disease with life
expectancies of a decade or two, to aggressive disease with a life
expectancy of only a few months, typically related to progression to
acute myeloid leukemia (AML).[1]
However, in all patients, MDS is a
clonal disease of hematopoiesis, and what determines slow progression
of marrow failure (without evolution to leukemia) in some patients, and
rapid progression to AML in others is only beginning to be understood.
More than forty somatic mutations in hematopoietic cells have been
described by now, and there is strong evidence that certain mutations,
for example, in ASXL1, RUNX1, SRSF2 or TP53, are associated with a more
rapid progression of the disease. Conversely, other mutations, for
example, in TET2, may be associated with slower disease evolution than
observed with wild-type TET2.[2,3]
Available data also suggest that
mutations in genes of the splicing machinery, occurring early in the
disease course, are the strongest “driver mutations”, and that
mutations, for example, in histone or DNA-modifying genes, in
transcription factors, such as TP53, and in kinase genes follow
later.[4] One clone may be dominant
early in the disease, but
follow-up, as described by Walter et al., will show the emergence of
sub-clones with new mutations, which may convey an altered
prognosis.[5,6] These observations
are important not only because of
the underlying disease mechanism and pathophysiology, but also because
a given treatment may prove to be more efficacious in the setting of
one mutation than in another and, in fact, may lead to the selection of
more resistant clones. These patterns are, of course, not unique to MDS
or AML; intra-tumor heterogeneity has been observed in many
malignancies.[7]
In addition to somatic mutations, numerous single nucleotide
polymorphisms (SNP) have been described that may affect the disease
process and may serve as predisposing factors for the development of
clonal disorders such as MDS.[8,9]
Further, recent data indicate that
even in the absence of mutations, dysregulation of expression of
various transcription factors, for example, TWIST1, and of microRNAs
(miRs) may be instrumental in disease evolution.[10-12]
In fact, it has
been suggested that miRs are the final regulators of tumor
progression.[13] While this
concept has added complexity to our
understanding of disease mechanisms, it is expected that these insights
will also lead to the development of novel therapies utilizing
recombinant RNA/DNA technology. While such therapy is currently “not
ready for prime time”, investigations are ongoing that use approaches
based on findings such as spliceosome mutations or miR dysregulation.
Current non-Tranplant Therapy for Mds
It is agreed upon that patients with a deletion of the long arm of
chromosome 5, del(5q), if they do have peripheral blood cytopenias, in
particular, anemias, should be treated with lenalidomide
(Revlimid).[14,15] The probability
of becoming transfusion independent
is about 65%, and many patients normalize their marrow, including
cytogenetics. On average transfusion independence extends over 2 to 2.5
years. However, even this remarkable treatment does not eradicate the
clone, which persists in stem cells.[16]
For other MDS patients, hypomethylating agents (HMA), i.e.
5-azacitidine (5-aza) or 2-deoxy-5-azacitidine, are standard therapy,
certainly for patients in IPSS categories intermediate-2 and high,
although patients with lower risk disease may benefit, by achieving
improved blood cell counts.[17-19]
Approximately 40% to 50% of patients
will have hematologic responses, leading to improved QOL. However, on
average these responses are sustained for only 9 to 10 months,
notwithstanding the fact that some patients may benefit for years.
Numerous other agents or combination therapy, for example, lenalidomide
combined with HMA, hold promise.[20]
However, none of these approaches
is curative. This raises several questions. If a patient is interested
in a curative approach, is initial treatment with HMA (or lenalidomide)
impacting the success of transplantation? If so, in a positive or
negative way? What should be the timing of HCT in a patient who is
responding to HMA? Has the patients acquired additional comorbidities
while receiving treatment with HMA (or other agents), while reflecting
on the possibility of transplantation? And how might those
comorbidities affect the decision for or against transplantation and
the success rate of HCT?
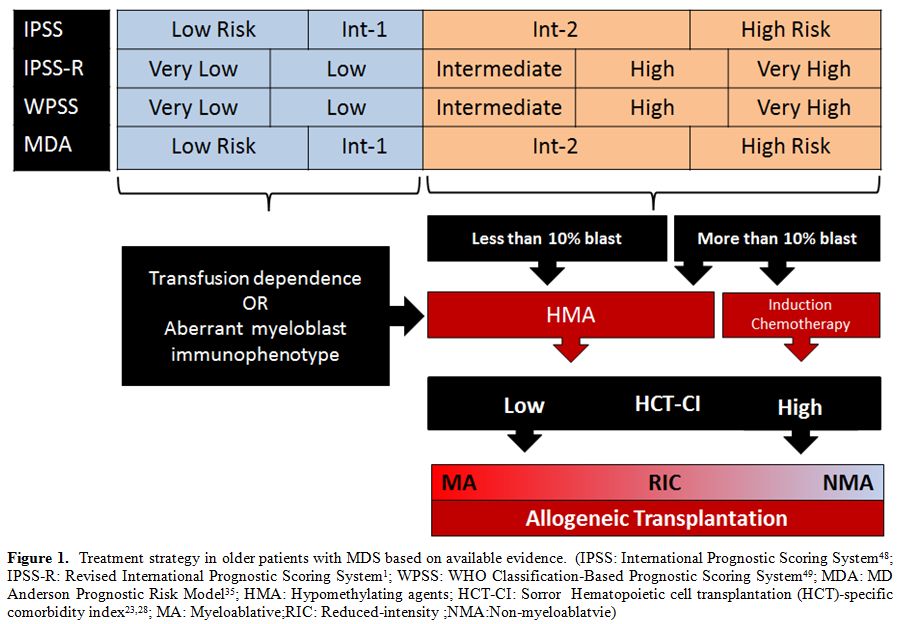 | Figure 1. Treatment strategy in older patients with MDS based on available evidence. (IPSS: International Prognostic Scoring System[48]; IPSS-R:Revised International Prognostic Scoring System[1]; WPSS:WHO Classification-Based Prognostic Scoring System[49]; MDA:MD Anderson Prognostic Risk Model[35]; HMA:Hypomethylating agents; HCT-CI:Sorror Hematopoietic cell transplantation (HCT)-specific comorbidity index[23,28]; MA:Myeloablative; RIC:Reduced-intensity; NMA:Non-myeloablatvie) |
Complicating Factors for Transplantation in Older Patients
At the time of diagnosis of MDS, patients are, on average, 70 to 75
years old. Some 10 or 15 years ago, one would not have considered HCT
for patients in this age range. A review of the literature shows,
however, that the median age of transplanted patients has increased
continuously over the past few decades. The database of the Center for
International Blood and Marrow Transplantation Research (CIBMTR) shows
a median patient age of 25 years in the 1980s, 39 in the 1990s, and 46
years over the past decade. Among patients transplanted between 2002
and 2009, 44% were older than 50 years, and the proportion of patients
older than 60 years increased from 8% to 17% between 2002 and 2011.[21]
Why the earlier
reluctance and why the current willingness to
transplant older patients? The often very intensive
conditioning
regimens used historically were poorly tolerated by older individuals,
presumably related to “biologic age”. However, it is clear from recent
studies that it is not so much chronological age but, rather, medical
issues, co-morbid conditions, more likely to be present with advanced
age that negatively impact outcome. Several scoring systems to assess
comorbidities and other factors that may affect transplant outcome have
been developed. Parimon et al.[22]
presented a predictive index
referred to as pre-transplantation Assessment of Mortality (PAM), based
primarily on donor type, disease risk, conditioning regimen, FEV1,
carbon monoxide diffusing capacity, serum creatinine, and serum alanine
aminotransferase concentrations, as well as age. Most of the studies
using the PAM have been carried out in patients transplanted following
high-intensity conditioning regimens, i.e. generally younger patients
and patients without comorbidities; a clear assessment and validation
of this test for older patients or patients conditioned with
reduced-intensity conditioning regimens remains to be done. Sorror and
colleagues developed the Hematopoietic Cell Transplantation Comorbidity
Index (HCT-CI), which considers only patient conditions, not other
factors that are part of the PAM.[23]
The HCT-CI includes various
cardiac, metabolic, cerebrovascular and hepatic parameters, but also
rheumatologic disorders, in addition to pulmonary dysfunction,
psychiatric disorders, and a prior history of solid tumors. This
scoring system has been applied to patients conditioned with
reduced-intensity “nonmyeloablative” conditioning regimens, and a clear
inverse correlation between the HCT-CI score and transplant outcome has
been shown.[24]
Most recently, Della Porta and colleagues presented data specifically
for patients with MDS whom they classified by using the revised IPSS (
IPSS-R), the HCT-CI (low/intermediate or high), the presence of
monosomal karyotype, refractoriness to induction chemotherapy, and
patient age (less than or greater than 50 years) for an MDS
transplantation risk index [TRI]).[25]
Their data on 519 patients
suggest that all these factors impact transplant outcome. Specifically,
the probability of long-term survival with a score of 0 or 1 was 70%,
whereas there was a median survival of only a few months (and no
long-term survival) among patients with a score of > 4, which
could
be reached, for example, with a high-risk HCT-CI, a high-risk IPSS-R,
and age > 50 years.
Of course, as shown by Naqui et al., comorbidities in patients with MDS
also impact negatively survival in non-transplanted patients.[26] In
other words, patients who would benefit most from definitive therapy
with HCT, are also the highest-risk patients when transplanted.
Therefore, detailed discussions with the patient in preparation for
transplantation are essential.
Koreth et al. analyzed data on 514 patients with de novo MDS, 60 to 70
years of age, and compared results obtained with HCT following
reduced-intensity conditioning (RIC) to results with best supportive
care (growth factor therapy or hypomethylating agents in patients with
intermediate-2/high-risk disease by IPSS).[27]
Patients with
low/intermediate-1 risk IPSS, had a life expectancy after
transplantation of 38 months, compared to 77 months for patients who
were not transplanted. A quality-adjusted life expectancy (QALE) and
sensitivity analysis did not favor the use of HCT in those patients.
Conversely, patients in intermediate-2/high-risk IPSS the life
expectancy was 36 months for transplanted, compared to 28 months for
non-transplanted patients, thereby favoring HCT. However, patients had
to survive for 3-4 years before this advantage of HCT became apparent.
What can an Older Patient Expect from Transplantation?
There are no randomized, prospective studies
comparing HCT
with non-transplant results in any population of patients, including
patients older than 60 or 70 years. Two such studies are currently
underway in Europe and the United States.
Published retrospective data have compared results in older and younger
patients with comparable diagnoses. For example, Sorror et al. provide
an analysis of transplant results in patients with various diagnoses
who were 60 to 64, 65 to 69, or 70 years of age and older, and
suggested that, based on likelihood ratio statistics using a Cox
regression model, there was no significant difference in survival
between those age brackets (P=0.18).[28]
Similar data have been
presented by others (see below). However, those results have to be
viewed critically. While statistical adjustments can be made in the
analysis, there is agreement that older patients were selected for
transplantation because they were considered “good candidates”,
i.e.
with no or few comorbidities, appearing younger than their stated age
with a high level of motivation and commitment. These patients,
therefore, do not necessarily reflect the average patient in that age
bracket in whom the diagnosis of a malignancy amenable to
transplantation is being made.
The Minnesota team analyzed results in 124 MDS (28%) and AML (72%)
patients 55-70 years of age who were conditioned with non-myeloablative
regimens and received transplants from HLA- matched related donors
(MRD) or were given umbilical cord blood (UCB).[29]
The overall
survival at 3 years was 37% for MRD and 31% for UCB transplants,
respectively. Acute (grades II-IV) and chronic GVHD rates were 38%-45%
and 26%-33% for MRD and UCB donors, respectively, and treatment–related
mortality at 2 years was 25%.
The European Bone Marrow Transplantation (EBMT) group reported results
in 1,333 patients who were 50 to 74 years old at the time of HCT and
carried the diagnosis of MDS or secondary AML.[30]
They were
transplanted from HLA-identical siblings (n=811) or from unrelated
donors (n=522). In this cohort, 883 patients were 50 to 60 years of
age, and 449 were older than 60 years. In this analysis, age was not a
significant risk factor for outcome. Relapse was determined by advanced
disease stage (P<0.01), and RIC regimen (P<0.01).
Non-relapse
mortality was determined by disease stage (P=0.01), the use of
unrelated donors (P=0.03), and the use of RIC regimens (P=0.03).
Thus, these data suggest that the selection of conditioning intensity,
which very likely considered patient age, was the most determining
factor for outcome.
The CIBMTR presented data on 1,080 patients transplanted between 1995
and 2005 following conditioning with RIC regimens.[31]
Among patients
with MDS or AML (in first remission), 2-year survival was 44%, 50%,
34%, and 36% for patients 40 to 54 years of age, 55 to 59 years, 60 to
64 years, or older than 64 years (P=0.05). Cytogenetics impacted
relapse and relapse-free survival. The 2-year overall survival was
determined by the pre-HCT performance status. Non-relapse mortality was
not significantly associated with chronologic age.
It remains challenging to define “biologic age”. Recent efforts have
considered frailty as a helpful parameter, suggesting an inverse
relationship between frailty and likelihood of success after HCT.[32]
Who and How?
Thus, the available data indicate that HCT is feasible in older
patients with MDS (and other myeloid diseases); however, as emphasized
above, no prospective trials are available. Therefore, how should
patients be selected and what transplant strategy should be pursued?
Clearly, disease and patient-associated risk factors, the tempo of the
disease, donor availability, among other factors, need to be
considered. Based on the results summarized here, patients with more
advanced disease, including IPSS intermediate-2, or high-risk patients
or with intermediate to very high-risk in the WPSS classification, and
patients in the intermediate to very high-risk categories as determined
by the IPSS-R scoring, should be considered for HCT. This does not
preclude, of course, transplantation for patients with lower risk under
particular circumstances, such as heavy transfusion dependence without
significantly elevated myeloblast counts and without high-risk
cytogenetics or other cytopenias, which per se would not place a
patient in a very high-risk category. We know, however, that in
patients with those characteristics the disease tends to progress more
rapidly, and carries a higher risk of transform into AML. Further, iron
accumulation associated not only with the disease itself but
particularly with the heavy transfusion load, may add comorbidity if
the transplant is delayed.[33]
Also, patients who by accepted criteria
have low-risk disease but have significantly aberrant immunophenotypes
of blast cells, as determined by flow cytometry,[34]
should have an
opportunity to discuss transplantation early in the course.
Investigators at the MD Anderson Cancer Center proposed several
additional risk scores, including a simplified MDS risk score that
considered poor performance status, older age, thrombocytopenia,
anemia, increased marrow blasts, leukocytosis, and high-risk
cytogenetics by IPSS criteria, and transfusion need as adverse risk
factors. Several studies have validated this scoring system, and
patients who have high scores by this assessment should probably be
assessed for transplantation if they are motivated, and the appropriate
support is available.[26,35]
Generally, patients with an HCT-CI score of >2 have experienced
considerably higher mortality post-transplant than patients with lower
scores. For patients with high HCT-CI scores even RIC or
“non-myeloablative” regimens as used currently may be associated with
unacceptable toxicity.
For patients with advanced disease, the challenge is two-fold: Since
older patients will be prepared for transplantation with RIC regimens,
providing a less cytotoxic component and a lesser debulking effect than
is achievable with high-intensity conditioning regimen, it appears
advisable (although no controlled data exist) to use debulking therapy
before transplantation. Several retrospective analyses have attempted
to determine the impact of pre-transplant therapy, particularly with
HMAs. It is premature to draw firm conclusions, again, because no
controlled studies are available. Clinical wisdom, however, holds that
a debulking attempt is indicated in patients with 5% myeloblasts or
more who are heading for a RIC transplant regimen. Classically,
induction-type chemotherapy has been used which historically has been
associated with mortality in the range of 10% or even higher. The
advent of HMAs offers an alternative. They are well tolerated, and 40%
to 50% of patients derive clinically relevant responses. Responding
patients, however, may be reluctant to proceed to HCT since they are
doing well and are not prepared to accept the potential risks
associated with HCT, but there are draw-backs.[36]
Prebet et al showed
that patients who received 5-azacitidine but their disease progressed
while on treatment, had a life expectancy of 5 – 6 months. HCT was the
only modality that offered any hope, but even so, the median survival
was only 1 to 1.5 years. On the other hand, patients who were taken off
treatment because they did not respond or did not tolerate the drug
and, therefore, went to transplantation, had a probability of about 40%
of becoming long-term survivors. Thus, if patients receive HMAs and
are interested in and are candidates for HCT, one should, presumably,
proceed to HCT while the patient is still responding, assuming a donor
is available.
Field et al., in a retrospective analysis, observed overall survival,
relapse-free survival, and relapse incidence at 1 year of 47%, 41%, and
20%, respectively, in patients who had received 5-azacitidine, compared
to 60%, 51%, and 32%, respectively, in patients who had not received
the drug before transplantation.[37]
As in other studies, however, the
selection of patients for treatment vs. no treatment was likely to be
biased.
One prospective study comparing 5-azacitidine to induction-type
chemotherapy in patients with advanced MDS who are candidates for HCT
was recently initiated by the FHCRC team with the objectives of
determining the response to either strategy, of determining what
proportion of patients with either approach could be brought to
transplantation, and assessing the impact on post-transplant
outcome.[38]
What is the Optimum Conditioning Regimen?
Even in younger patients the answer to this question is not clear. A
randomized trial comparing high-intensity and RIC regimens in patients
with MDS or AML conducted in the U.S. under the auspices of the BMT CTN
has recently been closed, and data should be forthcoming soon.[39] We
know from retrospective analyses that there are patients more than 60
or even 70 years of age who tolerate high-intensity conditioning
regimens as used in younger patients (for example, combinations of
busulfan and cyclophosphamide or fludarabine and busulfan).[40] The
general clinical policy is to condition patients older than 60 or 65
years with RIC regimens, although those “RIC” comprises regimens with
higher intensity than what is generally referred to as
“nonmyeloablative” (such as fludarabine + 2 Gy TBI). Those regimens
include combinations of fludarabine + melphalan or fludarabine +
reduced-dose busulfan (8–10 mg/kg, which can be administered in the
outpatient setting, favored by most patients and, conceivably, has a
cost-sparing effect as well.
However, the incidence of disease relapse after RIC in most such
studies has been higher than with high-intensity regimens; the
incidence of GVHD has been very similar, although the severity may be
lessened.[41] While disease
recurrence, observed in as many as 40% of
patients with high risk MDS, may return patients to a disease stage not
very different from the pre-HCT situation, GVHD clearly induces a new
problem, the classic “secondary disease”. Since first-line therapy
generally is with glucocorticoids, and older individuals often do not
tolerate glucocorticoids well, this scenario may lead to a downward
spiral of clinical deterioration. Patients may develop myopathy and
become progressively inactive, further enhancing the risk of
potentially fatal infections. New strategies are needed.
Very promising results have been obtained in recent years with
treosulfan-based regimens used in patients up to 65 years of age.[42]
Treosulfan metabolism differs from that of busulfan and is associated
with very little non-hematological toxicity. For patients who do not
have high-risk cytogenetics, 2-year relapse-free survival as high as
80% has been reported. Recent evidence suggest that the addition of 2
Gy TBI may improve results for patients with high-risk cytogenetics as
well, showing a 2-year relapse-free survival of about 65% (compared to
40% without incorporation of TBI), possibly related to a
radiosensitizing effect of treosulfan.[43]
Which Source of Stem Cells?
In recent years, the use of G-CSF-mobilized peripheral blood progenitor
cells (PBPC) has been favored because of more rapid engraftment, i.e.
shorter duration of pancytopenia, in particular neutropenia, and a more
potent anti-tumor (GVL) effect than observed with marrow cells.
However, a randomized study in patients receiving unrelated donor
transplants, similar to earlier studies with HLA-identical sibling
transplants, showed a higher incidence of chronic GHVD with PBPC, even
though this did not impact long-term survival.[44]
However, as stated
above, long-term steroid use (for GVHD) will create new problems,
particularly in these older patients and will affect the QOL.
Umbilical cord blood and HLA-haploidentical related donors offer
additional transplant options, greatly expanding the donor pool such
that a suitable donor/ source of stem cells is available for almost all
patients.[45] The use of cord
blood cells, in particular, has been
associated with relapse rates lower than seen, for example, with cells
from HLA matched unrelated donors; the GVHD incidence may not be
significantly different from that with HLA-matched donor cells.[46]
Some promising results have been reported also with HLA-haploidentical
donors, generally with the use of marrow as a source of stem cells. In
fact the incidence of GVHD tends to be lower than observed with
HLA-identical transplants, largely due to the effect of post-transplant
administration of cyclophosphamide ( days +3 and + 4).[47]
Summary and Conclusions
There has been considerable progress with the use of allogeneic HCT in
general, and in older patients and those with MDS in particular.
Regimen-related toxicity has been reduced with the development of a
range of different intensity conditioning regimens. Prevention and
treatment of GVHD, however, remain challenging tasks. Particularly
older patients, in whom GVHD and its treatment with glucocorticoids may
have a major impact, need to be fully aware of what they are facing
should they develop GVHD.
Nevertheless, all patients with MDS in their 60s or even early to
mid-70s should be offered an open discussion regarding an overall
treatment plan, including current standard therapy with transfusions,
growth factors or HMAs, as well as transplantation and long-term
outcome. A stem cell source can now be identified for almost all
patients, and the limiting factors in the decision-making process are
the patient’s comorbidities and their likely impact on transplant
outcome, the patient’s understanding of the procedure and long-term
effects, as well as the financial impact, not only on the patients
themselves but also on their families and, certainly, the long-term QOL.
Current and future research must further reduce treatment-related
morbidity and mortality. The transplant community must do better with
incorporating non-transplant modalities into the overall management of
patient with MDS, and some of these modalities such as HMAs or
immunotherapy must be investigated in well-designed post-transplant
studies.
Acknowledgments
We thank Helen Crawford and Bonnie Larson for help with manuscript
preparation.
References

[TOP]