Importance of Hyperbilirubinemia in Differentiation of Primary and
Secondary Hemophagocytic Lymphohistiocytosis in Pediatric Cases
Seval Ozen1, Alper Dai1, Enes Coskun1, Serdar Oztuzcu2, Sercan Ergun2, Elif Aktekin1, Sibel Yavuz1 and Ali Bay1,3
1 Gaziantep University Department of Pediatrics, Gaziantep, Turkey
2 Gaziantep University Department of Medical Biology, Gaziantep, Turkey
3 Gaziantep University Division of Pediatric Hematology Gaziantep, Turkey
Corresponding author: Ali Bay, Gaziantep Universitesi Tıp Fakultesi Cocuk Hastalıkları Klinigi, Gaziantep, Turkey. E-mail:
abay1968@yahoo.com
Published: November 1, 2014
Received: August 20, 2014
Accepted: October 3, 2014
Meditter J Hematol Infect Dis 2014, 6(1): e2014067, DOI
10.4084/MJHID.2014.067
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background and objective: Hemophagocytic
lymphohistiocytosis (HLH) is a life-threatening hyper-inflammatory
disease. It is difficult to differentiate between primary and secondary
HLH based on clinical findings at the onset of disease. We aimed to
find parameters that can help to differentiate primary and secondary
HLH at initial diagnosis especially for physicians working in
developing countries.
Patient and Method:
We retrospectively analyzed data of 38 HLH patients who were admitted
to the Pediatric Hematology Department of Gaziantep University between
January 2009 and December 2013.
Results:
Of 38 patients, 20 were defined as primary, and 18 were secondary HLH.
The average age of primary and secondary HLH patients was 31±9 and
81±14 months, respectively (p=0.03). We found consanguinity rates
significantly higher in primary HLH patients compared to secondary HLH
patients (p=0.03). We found that total and direct bilirubin levels
significantly increased in primary HLH patients compared to secondary
HLH patients (p=0.006, p=0.044). Also, CRP levels were found markedly
increased in secondary HLH patients compared to primary ones (p=0.017).
Conclusion:
We showed that cholestasis and hyperbilirubinemia findings of HLH
patients at the initial diagnosis should be considered in favor of
primary HLH, and an increased level of CRP should be considered in
favor of secondary HLH.
|
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening
hyperinflammatory disease caused by an uncontrolled and dysfunctional
immune response.[1] HLH has been categorized as primary or familial HLH
(FHLH), when there is a family history of HLH or known underlying
genetic defects. Reactive or secondary HLH occurs in the setting of
infection or underlying rheumatologic disorders or malignancy.[2] HLH
occurring in the setting of a rheumatological illness is commonly
referred to as macrophage activation syndrome (MAS).
However,
initial treatment should not govern disease classification (genetic or
acquired). However, information about the underlying genetic defect is
important for management because it will allow for an early search for
a stem cell donor.[3] Differentiation between primary and secondary
forms of HLH has become increasingly blurred together as new genetic
causes are identified.[4] In many developing countries, these genetic
tests are not performed, and blood had to be sent abroad for genetic
testing. Elongation of the process causes difficulties in the
follow-ups of these patients. It is difficult to differentiate between
primary and secondary HLH based on clinical symptoms, history of
infection, or the early clinical course at the onset of disease.[5]
Severity of disease and the identification of an infectious agent do
not differentiate between genetic and acquired forms of HLH. Age is
helpful to some extent a minority of children one year of age will have
acquired HLH, but older age does not reliably exclude genetic HLH.[6]
In
this study, we aimed to find parameters that can help to differentiate
primary and secondary HLH at initial diagnosis by comparing clinical
laboratory findings of 38 HLH patients followed in our clinic for last
four years. This procedure can be particularly useful for physicians
working in developing countries.
Patients and methods
From January 2009 to December 2013 we diagnosed 38 patients as HLH according to Diagnostic Guidelines for HLH 2004.[7]Patients
who were found to have a genetic abnormality and/or early-onset disease
(≤2 yr) with family history were considered as having familial HLH.
Patients whose genetic testing for UNC13D, PRF1, STX11, and STXBP2
revealed no genetic abnormality and who had no family history of HLH
were considered as having secondary HLH. 20
of 38 patients with genetic mutations detected or who had at least one
of the following conditions; family history or parental consanguinity,
persistence or recurrence of HLH, were classified as having primary
HLH. The remainder 18 patients without a genetic mutation detected and
who unmet the conditions mentioned above were classified as secondary
HLH. All
patients fulfilled at least five fundamental criteria of HLH at the
time of diagnosis, including fever, hepatosplenomegaly, bicytopenia
and/or pancytopenia, hypertriglyceridemia and/or hypofibrinogenemia,
hyperferritinemia, and hemophagocytosis in the bone marrow. Patients
were evaluated regarding with age, clinical findings, and laboratory
data using by descriptive statistics.
Statistical methods
SPSS.20
statistical software was used for the analysis. Student's t test and
Mann–Whitney U test were used, and p value less than 0.05 was evaluated
as statistically important
Results
A total of 38 HLH patients is included into this study. Of 38
patients, 20 were defined as primary, and 18 were secondary HLH.
Perforin, sytaxin, and munc13-4 mutations were detected in 6, 3, and 1
of primary HLH patients, respectively. The remaining ten patients were
considered to have primary HLH based on family history, age of onset
and recurrence of the disease, even if the genetic mutations were not
detected.
Out of 20 patients with primary HLH, 12 (60%) were
female, and 8 (40%) were male. Out of 18 patients with secondary HLH,
11 (61%) were female and 7 (39%) were male (39%). The average age of
primary and secondary HLH patients was 31±9 and 81±14 months,
respectively. Patients with primary HLH were significantly younger than
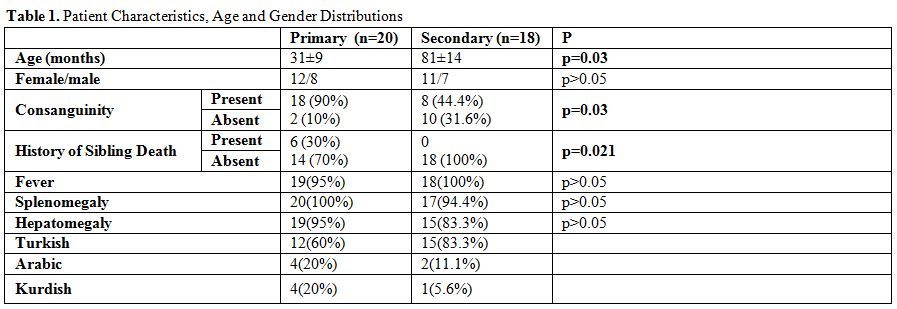
those with secondary HLH (p=0.03) (Table 1).
We
found consanguinity rates significantly higher in primary HLH patients
compared to secondary HLH patients (p=0.03). Also, the sibling death
history was present in 30% of primary patients but none in secondary
HLH patients. We did not detect any significant difference between
primary and secondary HLH patients when compared their clinical
findings such as fever, hepatomegaly, and splenomegaly. Also,
distribution of ethnicity can be seen on Table 1.
We
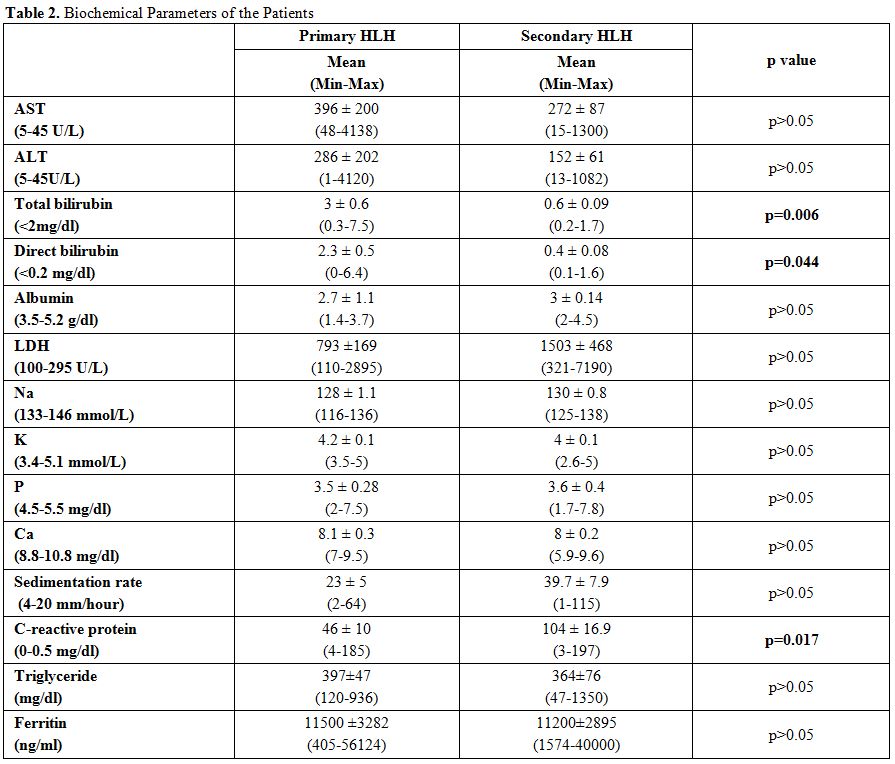
also compared the laboratory findings of HLH patients and; we found
that total and direct bilirubin levels significantly increased in
primary HLH patients compared to secondary HLH patients (p=0.006,
p=0.044) (Table 2). When we
took the cut-off level 1,3 mg/dl for total bilirubin level, we
calculated the sensitivity and specificity levels 60%, 95%
respectively. By the way when we took the cut-off level 0,8 mg/dl for
direct bilirubin levels, sensitivity and specificity levels were
calculated 60% and 95% respectively (Table 3).
Also, CRP levels were found markedly increased in secondary HLH
patients compared to primary ones (p=0.017). We calculated the
sensitivity and specificity levels 47% and 85% respectively, when we
took the cut-off level 98 mg/dl (Table 3).
We didn't find any significant difference between the two groups
by comparing the levels of WBC, hemoglobin, platelet, triglyceride,
fibrinogen, ferritin, transaminase, albumin, LDH, Na, K, P, Ca, PT,
INR, aPTT, and the sedimentation rate.
|
|
Table 1. Patient Characteristics, Age and Gender Distributions |
|
|
Table 2. Biochemical Parameters of the Patients |
|
|
Table 3. Sensitivity and Specificity Analysis of Total Bilirubin, Direct Bilirubin, and C-reactive protein |
Discussion
The
exact incidence or prevalence of HLH is not known. Based on the
available data, the incidence of HLH varies by geographic region.[8] It
has been reported to occur in anywhere from 1 of 50 000 live births in
Sweden[9] to 7.5 of 10 000 live births in Turkey;[10] this unusually
high reported prevalence is attributed to increased
consanguinity. Familial HLH comprises about 25% of all HLH, a number
that is more likely going to increase in coming years with the recent
boom in sequencing and genetic testing.[11,12] Acquired HLH, which
makes up majority of HLH in both children and adults, is not
associated with a known genetic defect by definition. The
hyper-inflammatory state is triggered by infectious, autoimmune, or
neoplastic conditions.[13]The
diagnostic criteria for HLH have been developed and updated in 2004 by
the FHL Study Group of the Histiocyte Society.[7] Making the diagnosis
of HLH is not sufficient. Identifying whether the patient has genetic
or acquired disease is also important for the management of the
disease. The treatment can be stopped in secondary HLH patients after
controlling the acute episode.[14] But in genetic, persistent and
recurrent HLH, continuation of treatment is recommended until SCT (stem
cell transplantation) is done.[15] Rapid detection of genetic disease
can provide an opportunity to start searching for an appropriate donor
quickly; also give the chance of starting an immediate treatment
and follow-up until a donor is available.[16]It
is important to predict whether patients have primary or secondary HLH
at the first admission in especially developing countries since the
investigation of HLA groups for stem cell transportation takes a long
time. In the literature, certain criteria for differentiation between
primary and secondary HLH have not been specified yet.[8,13] Impaired
NK cell cytotoxicity is a characteristic finding in FHL and
immunodeficiency syndromes with albinism; however, normal activity does
not exclude either.[6,17] Moreover, a decreased function of NK cells
has been observed in patients with acquired HLH, in patients with MAS,
and in close relatives of patients with FHL.[18] Recently,
flow cytometry has been used as a screening method to identify patients
with a genetic predisposition to HLH.[6,19] Intracellular stains
detecting perforin, SAP (X-linked lymphoproliferative syndrome
[XLP-1]), and XIAP (XLP-2) are available. Munc13-4 protein
expression in platelets has been reported as potential new rapid screen
for FHL-3 and was awaiting testing in a larger cohort.[20] Genetic
defects with impaired granule exocytosis (FHL 3-5, CHS, and GS-2) lead
to impaired translocation of the lysosome-associated membrane
glycoprotein CD107a to the cell surface, upon stimulation of NK cells
or cytotoxic T lymphocytes (CTLs). In
494 patients evaluated within a collaborative European study, the NK
degranulation assay clearly differentiated between patients with
defects in granule exocytosis and patients with acquired HLH or other
hereditary defects, such as perforin, SAP, or XIAP deficiency.[21] Once
these functional tests suggest a genetic basis for HLH, molecular
analysis should be followed, including for parents and siblings.Although
some articles suggesting that NK cell degranulation could be used for
differentiation of primary and secondary HLH have been published but
these methods are very hard to be used in developing countries. These
tests also require financial strength and special dyes in
flow-cytometry together with a certain experience. We investigated
whether differentiation of primary and secondary HLH is possible just
with clinical findings and laboratory tests which can be applied in
every facility easily. In our study, consanguinity and history of
sibling death rates were found significantly higher in primary HLH
patients as consistent with the literature when we compared the primary
and secondary HLH patients. The most intriguing finding in our study
was that total and direct bilirubin levels were increased in a manner
statistically significant in primary HLH patients compared to secondary
ones. This finding has not been reported in the literature before.
Recent study published by Japan Histiocytosis Study Group evaluated
prognostic factors of Epstein–Barr virus-associated hemophagocytic
lymphohistiocytosis in children.[22] They reported significantly higher
total bilirubin levels in non-survivors than in survivors. More
recently, it was indicated in a study reported from Vietnam that
hyperbilirubinemia on admission will be useful and could be a practical
predictor to determine high-risk HLH patients.[23] However, there was
no differentiation of primary and secondary HLH in those studies.Our
second finding is CRP levels found markedly increased in secondary HLH
patients compared to primary ones (p:0,017). In a study reported by
Stephan et al.,[24] CRP levels greater than 50 mg/L have been
associated with increased risk of infection and overall mortality in
HLH patients with underlying autoimmune disorders. Thus, ESR or CRP may
be used as indices of disease severity, but care must be taken to
identify coincident inflammatory insults such as infection or
autoimmune disease. There are no patients with complicating bacterial
infection in our secondary HLH group. There are little data showing
increased level of CRP in HLH patients. In studies conducted on adult
patients mostly, CRP levels were detected as higher in HLH group when
compared to not having HLH. In pediatric and adult age groups, there
were no studies comparing CRP levels between the primary and secondary
HLH. A possible cause of CRP increase is that stimulants needed for the
development of HLH should be stronger due to the absence of a genetic
defect in secondary HLH patients. Therefore, this may be speculated as
like that CRP level is higher during more severe inflammation
developing as a result of strong stimulants. Therefore, this situation
may explain the increase of CRP in secondary HLH.In
our study, we investigated the criteria that can be used in the
differentiation of FHL and secondary HLH for the patients especially in
countries not having advanced laboratory facilities. As a result, we
showed that the cholestasis and hyperbilirubinemia, found in HLH
patients at the initial diagnosis, should be considered in favor of
primary HLH. On the contrary, an increased level of CRP should be
considered in favor of secondary HLH. These data should be confirmed
being our study conducted for a limited number of patients in a single
center.
References
- Bode SF, Lehmberg K, Maul-Pavicic A, et al. Recent
advances in the diagnosis and treatment of hemophagocytic
lymphohistiocytosis. Arthritis Res Ther. 2012;14:213.
http://dx.doi.org/10.1186/ar3843

- Bay A,
Bosnak V, Leblebisatan G, et al. Hemophagocytic lymphohistiocytosis in
2 pediatric patients secondary to hepatitis A virus infection. Pediatr
Hematol Oncol. 2012;29:211-4.
http://dx.doi.org/10.3109/08880018.2012.666783
- Filipovich
AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr
Opin Allergy Clin Immunol. 2011;11:512-516.
http://dx.doi.org/10.1097/ACI.0b013e32834c22f5
- Johnson
TS1, Villanueva J, Filipovich AH, Marsh RA, Bleesing JJ. Contemporary
diagnostic methods for hemophagocytic lymphohistiocytic disorders. J
Immunol Methods. 2011;364:1-13.
http://dx.doi.org/10.1016/j.jim.2010.11.006
- Janka
GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev
Med. 2012;63:233-246.
http://dx.doi.org/10.1146/annurev-med-041610-134208
- Janka
GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and
treatment. Hematology Am Soc Hematol Educ Program. 2013;2013:605-11. http://dx.doi.org/10.1182/asheducation-2013.1.605
- Henter
JI, Horne A, Aric_oM, et al. HLH-2004: Diagnostic and therapeutic
guidelines for hemophagocytic lymphohistiocytosis. Ped Blood Cancer
2007;48:128–131. http://dx.doi.org/10.1002/pbc.21039
- Tothova
Z, Berliner N. Hemophagocytic Syndrome and Critical Illness: New
Insights into Diagnosis and Management. J Intensive Care Med published
online 8 January 2014
- Henter JI, Elinder
G, Soder O, Ost A. Incidence in Sweden and clinical features of
familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand.
1991;80:428-435.http://dx.doi.org/10.1111/j.1651-2227.1991.tb11878.x
- Gurgey
A, Gogus S, Ozyurek E, et al. Primary hemophagocytic
lymphohistiocytosis in Turkish children. Pediatr Hematol Oncol.
2003;20:367-371. http://dx.doi.org/10.1080/08880010390203891
- Rohr
J, Beutel K, Maul-Pavicic A, et al. Atypical familial hemophagocytic
lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with
primary immunodeficiency diseases. Haematologica. 2010;95:2080-2087. http://dx.doi.org/10.3324/haematol.2010.029389
- Pagel
J, Beutel K, Lehmberg K, et al. Distinct mutations in STXBP2 are
associated with variable clinical presentations in patients with
familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood
2012;119(25):6016-6024. http://dx.doi.org/10.1182/blood-2011-12-398958
- Bay
A, Coskun E, Oztuzcu S, Ergun S, Yilmaz F, Aktekin E. Evaluation of the
Plasma Micro RNA Expression Levels in Secondary Hemophagocytic
Lymphohistiocytosis. Mediterr J Hematol Infect Dis 2013 Nov
4;5(1):e2013066.
- Rouphael NG,
Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections
associated with haemophagocytic syndrome. Lancet Infect Dis 2007; 7:
814–822. http://dx.doi.org/10.1016/S1473-3099(07)70290-6
- Horne
A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell
transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol.
2005;129:622-630.
http://dx.doi.org/10.1111/j.1365-2141.2005.05501.x
- Chandrakasan
S, Filipovich AH. Hemophagocytic lymphohistiocytosis: advances in
pathophysiology, diagnosis, and treatment. J Pediatr. 2013 ;163:1253-9. http://dx.doi.org/10.1016/j.jpeds.2013.06.053
- Schneider,
E.M., Lorenz, I., Muller-Rosenberger, M., Steinbach, G., Kron, M.,
Janka-Schaub, G.E. Hemophagocytic lymphohistiocytosis is associated
with deficiencies of cellular cytolysis but normal expression of
transcripts relevant to killer-cell-induced apoptosis. Blood 2002:100,
2891-8. http://dx.doi.org/10.1182/blood-2001-12-0260
- Villanueva
J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, Grom AA.
Natural killer cell dysfunction is a distinguishing feature of systemic
onset juvenile rheumatoid arthritis and macrophage activation syndrome.
Arthritis Res Ther. 2005;7:30-7. http://dx.doi.org/10.1186/ar1453
- Lehmberg K, Ehl S. Diagnostic
evaluation of patients with suspected haemophagocytic
lymphohistiocytosis. Br J Haematol. 2013;160:275-287.
http://dx.doi.org/10.1111/bjh.12138
- Murata
Y, Yasumi T, Shirakawa R, et al. Rapid diagnosis ofFHL3 by flow
cytometric detection of intraplatelet Munc13-4 protein. Blood.
2011;118:1225-1230.
http://dx.doi.org/10.1182/blood-2011-01-329540
- Bryceson
YT, Pende D, Maul-Pavicic A, et al. A prospective evaluation of
degranulation assays in the rapid diagnosis of familial hemophagocytic
syndromes. Blood. 2012;119:2754-2763.
http://dx.doi.org/10.1182/blood-2011-08-374199
- Kogawa
K, Sato H, Asano T, Ohga S, Kudo K, Morimoto A, Ohta S, Wakiguchi H,
Kanegane H, Oda M, Ishii E. Prognostic factors of Epstein-Barr
virus-associated hemophagocytic lymphohistiocytosis in children: Report
of the Japan Histiocytosis Study Group. Pediatr Blood Cancer.
2014;61:1257-1262. http://dx.doi.org/10.1002/pbc.24980
- Dao
AT, Luong VT, Nguyen TT, Huynh QT, Phan TT, Lam MT, Ngoma AM, Koriyama
C. Pediatr Hematol Oncol. Risk Factors for Early Fatal Outcomes Among
Children with Hemophagocytic Lymphohistiocytosis (HLH): A
Single-Institution Case-Series in Vietnam. Pediatr Hematol Oncol.
2014;31:271-81.
- Stéphan JL1, Koné-Paut
I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM. Reactive
haemophagocytic syndrome in children with inflammatory disorders. A
retrospective study of 24 patients. Rheumatology (Oxford). 2001;40:1285-92. http://dx.doi.org/10.1093/rheumatology/40.11.1285