Received: May 22, 2014
Accepted: September 26, 2014
Meditter J Hematol Infect Dis 2014, 6(1): e2014071, DOI 10.4084/MJHID.2014.071
This article is available on PDF format at:

MR El-Shanshory1, AA Hagag1, SS Shebl1, IM Badria1, AH Abd Elhameed2, ES Abd El-Bar2, Y Al-Tonbary3, A Mansour3, H Hassab4, M Hamdy5, M Elalfy6, L Sherief7 and E Sharaf8
1 Pediatric Department, Tanta University, Egypt
2 Clinical Pathology Department, Tanta University, Egypt
3 Pediatric Departments of Mansoura University
4 Pediatric Departments of Alexandria University
5 Pediatric Departments of Cairo University
6 Pediatric Departments of Ain Shams University
7 Pediatric Departments of Zagazeg University
8 Pediatric Departments of Sohag University
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Background: The
molecular defects resulting in a β-thalassemia phenotype, in the
Egyptian population, show a clear heterogenic mutations pattern.
PCR-based techniques, including direct DNA sequencing are effective on
the molecular detection and characterization of these mutations. The
molecular characterization of β-thalassemia is necessary for carrier
screening, genetic counseling, and to offer prenatal diagnosis. The aim of the work: was to evaluate the different β-globin gene mutations in two hundred β-thalassemic Egyptian children. Subjects and Methods:
This study was carried out on two hundred β-thalassemic Egyptian
children covering most Egyptian Governorates including 158 (79%)
children with thalassemia major (TM) and 42 (21%) children with
thalassemia intermedia(TI). All patients were subjected to meticulous
history taking, clinical examination, complete blood count, hemoglobin
electrophoresis, serum ferritin and direct fluorescent DNA sequencing
of the β-globin gene to detect the frequency of different mutations. Results:
The most common mutations among patients were IVS I-110(G>A) 48%,
IVS I-6(T>C) 40%, IVS I-1(G>A) 24%, IVS I-5(G>C)10%, IVS
II-848 (C>A) 9%, IVS II-745(C>G) 8%, IVS II-1(G>A) 7%,
codon"Cd"39(C> T) 4%, -87(C>G) 3% and the rare mutations
were: Cd37 (G>A), Cd8 (-AA), Cd29(-G), Cd5 (-CT), Cd6(-A),
Cd8/9(+G), Cd 106/107(+G), Cd27(C>T), IVS II-16(G> C), Cd 28
(-C), Cap+1(A>C), -88(C>A), all of these rare mutations were
present in 1%. There was a considerable variation in phenotypic
severity among patients resulting from the interaction of different β°
and β+mutations. Furthermore, no genotype-phenotype
association was found both among the cases with
thalassemia major and the cases with thalassemia intermedia. Conclusion:
Direct DNA sequencing provides insights for the frequency of different
mutations in patients with β- thalassemia including rare and/or unknown
ones. The most common mutations in Egyptian children with beta
thalassemia were IVS I-110(G>A) 48%, IVS I-6(T>C) 40%, IVS
I-1(G>A)24% , IVS I-5(G>C)10%, IVS II-848 (C>A) 9%, IVS
II-745(C>G) 8%, IVS II-1(G>A) 7%. |
Introduction
Thalassemia syndrome is the most common single gene disorder.[1] It
is an autosomal recessive hereditary anemia due to mutations that
reduce (β+) or abolish (βº) synthesis of β-globin chains of hemoglobin
tetramer, which is made of two alpha and two beta globin chains (α2
& β2) required for HbA formation.[2] The disease is very
heterogeneous at the molecular level, with more than 300 different
molecular defects defined to date.[3]
As in many Mediterranean
countries, β-thalassemia is a major public health problem in Egypt. The
position of Egypt in the center of the Middle East, contiguous with the
Mediterranean countries, has facilitated genetic admixture of Egyptians
with several populations of diverse geographic and ethnic origins.[4]
It
has been estimated that 1000 children out of 1.5 million live births
are born annually with thalassemia major.[5] In multicenter studies,
the carrier rate in Egypt has been reported to be in the range of
9%-10%.[4]
Treatment of β-thalassemia, albeit more and more
available, remains a significant drain on the country's resources.
Regular blood transfusions in combination with iron chelation have
remarkably increased the life-span of patients with β-thalassemia.[6]
However,
iron-related complications, including life-threatening ones such as
heart disease, are still common. A prevention program would be useful
to overcome these problems, but it requires a preliminary knowledge of
the most common β-globin mutations among the population.[7]
DNA
sequencing as availability of this method and standardization of this
technique in the country can help in choosing the best strategy for
molecular diagnosis with the possibility to detect rare mutations in
the area.[8]
The present work aimed to evaluate the different
β-globin gene mutations in two hundred of Egyptian children with
β-thalassemia by direct DNA sequencing to be taken in consideration of
prevention program of β-thalassemia.
Subjects and methods
This study was conducted on 200 cases of children with β-thalassemia
including 158 children with thalassemia major and 42 children with
thalassemia intermedia. An informed consent was obtained from all
parents of children before enrollment in the study. The study was
approved by the Ethical Committee of Tanta University.
These
children came from most of Egyptian Governorate with a random selection
from thousand cases (Alexandria, Cairo, Al-Gharbiyah, Al Manofia, Kafr
El Sheikh, Sohag, Al-Fayoum, Al Kaluobia, Port Said, Al-Dakhlia,
Domiat, Al Jizaz, and Al-Beheira). The rest traces from other
Governments.
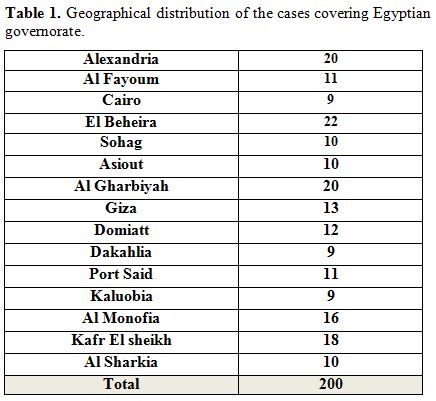 |
Table 1. Geographical distribution of the cases covering Egyptian governorate |
 |
Figure 1. Map showing geographic distribution of different governorates in Egypt |
All
patients were subjected to meticulous history taking with reference to
positive consanguinity and clinical evaluation of all body systems. All
affected patients were clinically classified into thalassemia major or
intermedia with consideration to: age of disease onset, age of first
transfusion, frequency of blood transfusion, hemoglobin level,
hepatosplenomegaly, facial and growth affection.[8]
Routine
hematological investigations e.g.: complete blood count using ERMA
PCE-210 N cell counter, reticulocyte count, Hb electrophoresis using
cellulose acetate in a tris EDTA borate buffer at PH 8.4 (Helena
Laboratories, Beaumont, TX, USA), serum ferritin levels using Monobind
Inc ELISA Microwells kit (lake Forest, CA 92630, USA).
Children
with beta thalassemia major and intermedia were studied with DNA
sequencing: DNA extraction and purification was performed from whole
blood collected in EDTA-containing tubes, by using a QIA amp DNA blood
mini kit (Qiagen, Hilden, Germany CA. No. 51104), according to the
manufacturer`s instruction.
The PCR amplification products
of each sample were applied to gel electrophoresis (2% agarose gel
stained with ethidium bromide) and visualized under UV illumination
(Biometra Germany). The samples were detected as a clear, sharp,
distinct band at the specific molecular weight (550 bp, for Hemoglobin
subunit beta-1 (HBB1), 650 bp for Hemoglobin subunit beta-2) (HBB2).The
positive PCR products were then purified by PCR purification columns,
using QIA QuickR PCR Purification kit
(Qiagen, Hilden, Germany cat. No. 28104). Then subjected to cycle
sequencing PCR using fluorescent dyes (Applied Biosystems, Foster City,
CA, USA).
Following the cycle sequencing PCR, the samples were
then purified to remove low molecular weight components like
nucleotides and buffer salts, using CENTRI-SEP columns (cat. No.
CS-901). The cycle Sequence products were then analyzed with an
automated sequencer (ABI PRISMTM 310
Genetic Analyzer). Finally: Interpretations of the results via SeqScape
software version 2.7 Applied Biosystem.[9] The Primer sequences are not
available from the manufacturer”.
Statistical Analysis.
Data were analyzed using SPSS version 20. Data were expressed as mean ±
standard deviation for quantitative variables, number and percentage
for qualitative ones with the use of Chi-square, ANOVA tests. P value
< 0.05 was considered to be statistically significant.
Results
There were no significant differences between patients with
thalassemia major and thalassemia intermedia regarding age, sex, family
history of thalassemia, consanguinity (fifty-five percent of
thalassemic patients had positive consanguinity, and 40% had a positive
family history of thalassemia (presence of one brother or
sister suffering from thalassemia), weight, height and body mass
index (BMI).
Pallor and jaundice were the most common presenting
symptoms while hepatomegaly and splenomegaly were the most common
presenting signs in patient's group.
The age of 1st transfusion in
studied patients ranged from 4-72 months, with a mean age of first
transfusion of 16.52 ± 5.96 months, and interval of transfusion ranged
from 2-24 weeks with mean interval of 4.09 ± 2.29 weeks. There were
significantly lower red blood cells (RBCs), hemoglobin (Hb), and
significantly higher reticulocytes, platelets and white blood cells
(WBCs) in patients with thalassemia major compared with patients with
thalassemia intermedia with no significant differences between patients
with thalassemia major and thalassemia intermedia as regard mean
corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) (Tables 2,3).
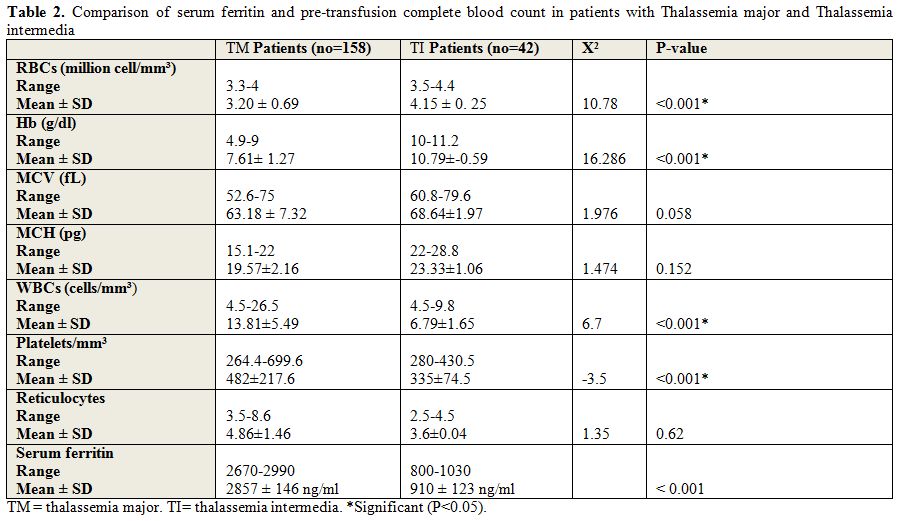 |
Table 2. Comparison of serum ferritin and pre-transfusion complete blood count in patients with Thalassemia major and Thalassemia intermedia |
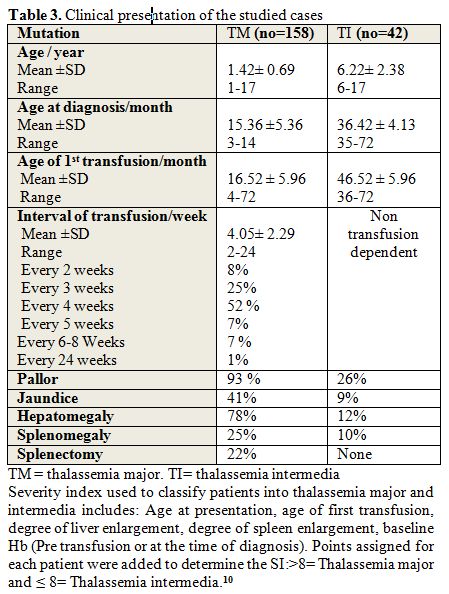 |
Table3. Clinical presentation of the studied cases |
There
was significantly lower total iron binding capacity, and significantly
higher serum ferritin and serum iron, in patients with thalassemia
major compared with patients with thalassemia intermedia (serum
ferritin was 2857 ± 146 ng/dl in thalassemia major versus 910 ±
123 ng/dl in thalassemia intermedia with p value < 0.001) .
Globin mutations are presented in table 4 including common and rare mutations as follow:
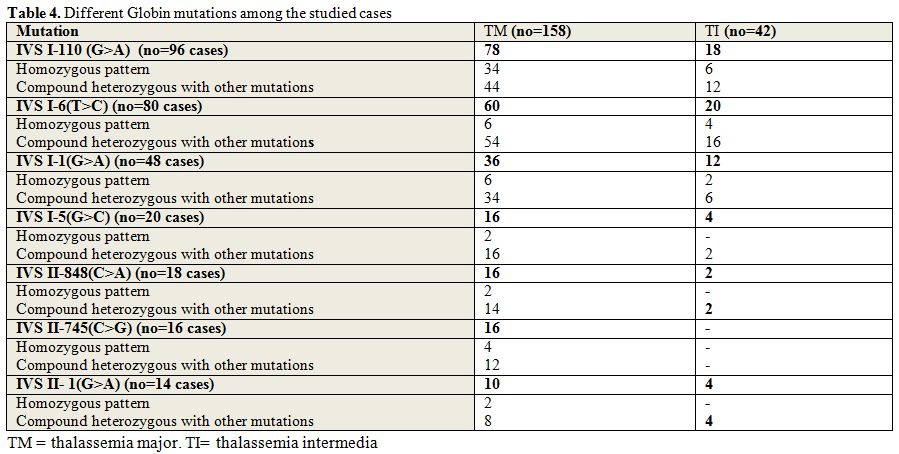 |
Table 4. Different Globin mutations among the studied cases |
Common mutations:
The most common mutations among patients were IVS I-110(G>A), which
was present in 96 cases out of two hundred (48%), homozygous pattern
was present in 40 cases of them; compound heterozygous with other
mutations was present in 56 cases; IVS I-6(T>C), which was present
in 80 cases (40%), 10 of them were homozygous and 70 were compound
heterozygous; IVS I-1(G>A), which was present in 48 cases (24%), 8
of them were homozygous, and 40 were compound heterozygous; IVS
I-5(G>C), which was present in 20 cases (10%), 2 of them was
homozygous, and 18 were compound heterozygous; IVS II-848(C>A),
which was present in 18 cases (9%), 2 of them was homozygous, and 16
were compound heterozygous; IVS II-745(C>G), which was present in 16
cases (8%), 4 of them were homozygous, and 12 were compound
heterozygous; IVS II- 1(G>A), that was present in 14 cases
(7%), 2 of them was homozygous, and 12 were compound heterozygous;
Cd39(C>T), which was present in 8 cases (4%), 2 of them was
homozygous, and 6 were compound heterozygous; -87(C>G), which was
present in 6 cases (3%), all of them were compound heterozygous.
Rare mutations:
Cd37 (G>A), Codon 8 (-AA), Cd29(-G), Codon5 (-CT), cd6(-A),
Cd8/9(+G), Cd 106/107(+G), Cd27(C>T), IVS II-16(G> C), Codon 28
(-C), Cap+1(A>C), -88(C>A) all of these rare mutations were
present in 1% all of them were compound heterozygous, except for Cd37
(G>A),and IVS II-16(G> C) were homozygous.
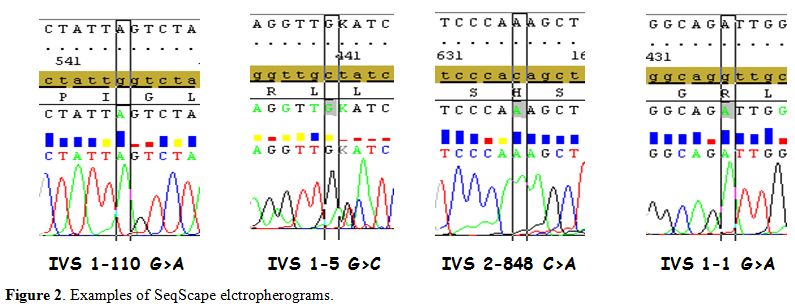 |
Figure 2. Examples of SeqScape elctropherograms |
Discussion
β-Thalassemia is the most common genetically inherited hemoglobin disorder in Egypt.[11]
The molecular defects resulting in a β-thalassemia phenotype, in the
Egyptian population show a clear heterogenic pattern. Many studies have
embarked on the molecular detection and characterization of these
mutations, using a wide array of the available techniques with
successful detection of both known and unknown mutations. PCR-based
techniques, including direct DNA sequencing are effective with some
limitations about the time, effort and high cost to reach a final
diagnosis.[12]
The aim of this work was to
evaluate the different β-globin gene mutations in two hundred
β-thalassemic Egyptian children.
In the present study, the
most common mutations among patients were IVSI-110(G>A) which were
present in 96 cases out of two hundred (48%), and IVSI-6(T>C) was
present in 80 cases (40%), then IVSI-1(G>A) in 48 cases
(24%), IVSI-5(G>C) in 20 cases (10%), IVSII-848(C>A) in 18
cases (9%), IVSII-745(C>G) in 16 cases (8%), IVSII-1(G>A) in 14
cases (7%), Cd39(C>T) in 8 cases (4%), -87(C>G) in 6 cases (3%).
These results were in agreement with Hussein et al., 2007,[13]
who found 12 different mutations in patients from Suez Canal region;
the most frequent mutations were IVSI-110 (G→A) (31.4%),
IVSI-1(G→A)(17.6), IVSI-6(T→C)(17.6%), -87(C>G)(7.8%),
IVSII-1(G>A)(5.9%), IVSII-745(C> G)(5.9%).
This study was in accordance with Kaddah et al., 2009,[14]
who reported that the most common seven genetic mutations of the
β thalassemia evaluated in Egyptian studies were IVSI-6, IVSI-110,
IVSII-1, IVSII-745, IVSI-1, -87 and codon 39. Also Settin et al., 2006[15]
stated that three abundant mutations were found accounting for a total
71.25% of all mutations; these 3 mutations were IVS I-110 (G→A), IVS
I-6 (T→C) and IVS I-1 (G→A) representing 37.5%,17.5% and 16.25%
respectively.
Jiffri et al., 2010[16] in another
specified study of Upper Egypt agreed with our study finding that the
most frequent mutation was IVS-I-110 (G→A) (57%). The IVS-I-110,
IVS-I-6 (T→C) and IVS-I-1 (G→A) mutations accounted for 87% of
mutations in the β-thalassemia.
Consistent with this study Omar et al., 2005,[17]
in Alexandria, reported the most common mutations are IVSI-110(62%)
followed by IVSI-6(7%) and IVSI-1 (4%), other mutations IVSII-1 &
Cd-39 are not found in any of the studied patients.
On the other hand El-Gawhary et al., 2007,[18]
reported that IVSI-6 is more frequent than IVSI-110, but their study
covered Fayoum in Upper Egypt, Cairo, Alexandria and Tanta in Lower
Egypt and the Nile Delta. The proportion of IVS-I-6 (T→C) was 36.3% and
of IVSI-110 (G→A) 25.8%.
Rare mutations in our study:
Cd37(G>A), Cd8(-AA), Cd29(-G), Cd5(-CT), Cd6(-A), Cd8/9(+G),
Cd106/107(+G), Cd27(C>T), IVSII-16(G> C), Cd28(-C),
Cap+1(A>C), -88(C>A) all of these rare mutations were present in
1%.
There was a considerable variation in phenotypic severity
among patients resulting from interaction of different β°and
β+mutations, 158 (79%) cases were thalassemia major (TM) and 42 (21%)
were thalassemia intermedia (TI). This result was in agreement with
Nadkarni et al., 2007,[10] and Omar et al., 2005.[17]
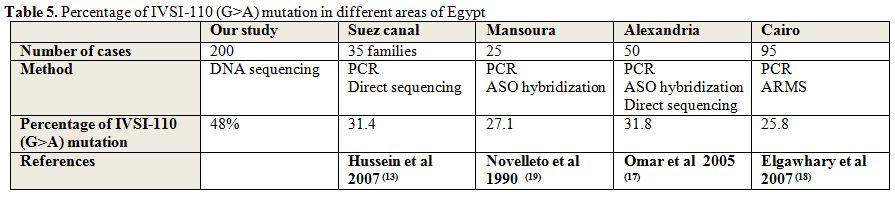 |
Table 5. Percentage of IVSI-110 (G>A) mutation in different areas of Egypt |
Conclusion
Direct DNA sequencing provides insights for the frequency of different mutations in patients with β- thalassemia including rare and /or unknown ones. The most common mutation in Egyptian children with beta thalassemia were IVS I-110(G>A) 48%, IVS I-6(T>C) 40%, IVS I-1(G>A) 24%, IVS I-5(G>C) 10%, IVS II-848 (C>A) 9%, IVS II-745(C>G) 8%, IVS II-1(G>A) 7%.
References

[TOP]