Received: November 2, 2014
Accepted: November 20, 2014
Meditter J Hematol Infect Dis 2015, 7(1): e2015001, DOI 10.4084/MJHID.2015.001
This article is available on PDF format at:

Elena Maino1, Anna Maria Scattolin1, Piera Viero1, Rosaria Sancetta1, Anna Pascarella1, Michele Vespignani1 and Renato Bassan1
1 Hematology and Bone Marrow Transplant Unit, Ospedale dell’Angelo e SS. Giovanni e Paolo, Mestre-Venezia, Italy
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract The introduction
of newer cytotoxic monoclonal antibodies and chimeric antigen receptor
modified T cells is opening a new age in the management of B-lineage
adult acute lymphoblastic leukemia. This therapeutic change must be
very positively acknowledged because of the limits of intensive
chemotherapy programs and allogeneic stem cell transplantation. In
fact, with these traditional therapeutic tools the cure can be achieved
in only 40-50% of the patients. The failure rates are particularly high
in the elderly, in patients with post-induction persistence of minimal
residual disease and especially in refractory/relapsed disease. The
place of the novel immunotherapeutics in improving the outcome of adult
patients with B-lineage acute lymphoblastic leukemia is
reviewed.
|
Introduction
Adult acute lymphoblastic leukemia (ALL) is biologically
heterogeneous and can be subdivided into several clinico-prognostic
entities.[1] The primary distinction is between B-cell and T-cell
precursor (BCP, TCP) ALL, and in the former group between Philadelphia
chromosome/BCR-ABL (Ph) positive and Ph- ALL. The overall outcome of
adults with ALL is inferior to that of childhood ALL. Basically,
survival is strictly related to a complete remission (CR) achieved
early on, which is followed by an effective consolidation/maintenance
therapy, in standard-risk patients (SR) and, an allogeneic stem cell
transplantation (SCT), in high-risk (HR) patients.[2,3] In adolescent
and adult patients with Ph- ALL in an age range between 15-18 and 60-65
years, the CR rate is 90% and, the overall survival (OS) rate is 40-50%
at 3-5 years, with significant differences among age and risk
groups.[4,5] In Ph+ ALL, results are suboptimal too despite the
improvement due to the introduction of tyrosine kinase inhibitors.[6]
In Ph- ALL, better OS, and disease-free survival (DFS) rates are
increasingly reported using pediatric-inspired schedules, at least in
patients aged up to 40-50 years.[7] The outcome is worse in patients
older than 55 years, with smaller proportions of long-term
survivors.[8] Moreover during CR induction about 5% of the patients
succumb to early complications, mainly infectious, and the risk of
non-relapse mortality is still rather high after an allogeneic SCT (15%
on the average). Overall, the common perception is that treatment
intensity cannot be increased any further beyond this point in adult
patients, without incurring into unacceptably high rates of
treatment-related toxicity and mortality. Instead, new alternative
therapeutics should be developed with a view of reducing the toxicity
burden other than improving the antileukemic efficacy of available
antileukemic programs. In addition, the relapse rate in adult ALL
remains high and salvage therapy is at present unsatisfactory, with an
effective rescue rate of 10-20% in most studies.
The most recent therapeutic innovations are represented by newer
monoclonal antibodies (MoAb) and the chimeric antigen receptor (CAR)
modified T cells. These new, highly selective weapons target specific
ALL cell antigens and would exhibit an improved activity versus
toxicity ratio compared to chemotherapy or transplantation. In
addition, they could be used sequentially or in combination with either
treatment modality, to potentiate the overall treatment efficacy. Thus
far, MoAb-based therapy and CAR T cell therapy were developed mainly
for B-lineage but not T-lineage ALL. They have been utilized in all
B-lineage subsets (BCP and mature B/Burkitt ALL; Ph- and Ph+ ALL), and
demonstrated considerable activity in relapsed/refractory disease (R/R
ALL). Therefore, they need to be exploited in untreated ALL, especially
in high-risk subsets such as the elderly and the patient with high
post-induction levels of minimal residual disease (MRD). Here we review
the evidence supporting the use of therapeutic MoAb and CAR T cells in
BCP ALL. Additional data can be found elsewhere.[9-11] Results from
childhood studies will be reported whenever appropriate to illustrate
specific points of interest.
Modern Immunotherapy with Monoclonal Antibodies
The challenge of novel immunotherapeutics is to improve
survival without increasing toxicity. With MoAbs, the different and
manageable toxicity profile only occasionally overlaps or worsens that
associated with chemotherapy and SCT. For instance, mucositis and
gastrointestinal toxicity, usually of high concern with intensive
chemotherapy and SCT, are not typical of MoAb therapy. The apparent
lack of cross-resistance with standard antileukemic drugs constitutes a
further theoretic advantage. The third major issue is whether MoAb
therapy might substitute, at least partially, for some intensive
chemotherapy elements and/or SCT in patients in CR1. Prospective
clinical trials should address this most important topic.
ALL cells express several membrane antigens. The ideal therapeutic
target should be consistently expressed in every ALL subset, by all
blast cells, at high intensity, be stable upon MoAb challenging and
play a crucial role in metabolic events. At present, no MoAb satisfies
all these requirements and a target expression of 20% out of the entire
ALL cell population is considered enough to start a MoAb trial with
some chance of success.
According to their structural characteristics and mechanism of action,
MoAbs for ALL therapy belong to three major categories: naked
antibodies, T-cell engaging bispecific single-chain (BiTE®) antibodies,
and immunoconjugates/immunotoxins. The several trials launched with the
most representative and therapeutically promising MoAb’s, with or
without associated chemotherapy, are summarized in Figure 1 (frontline
studies) and Figure 2
(studies in R/R ALL) and detailed below.
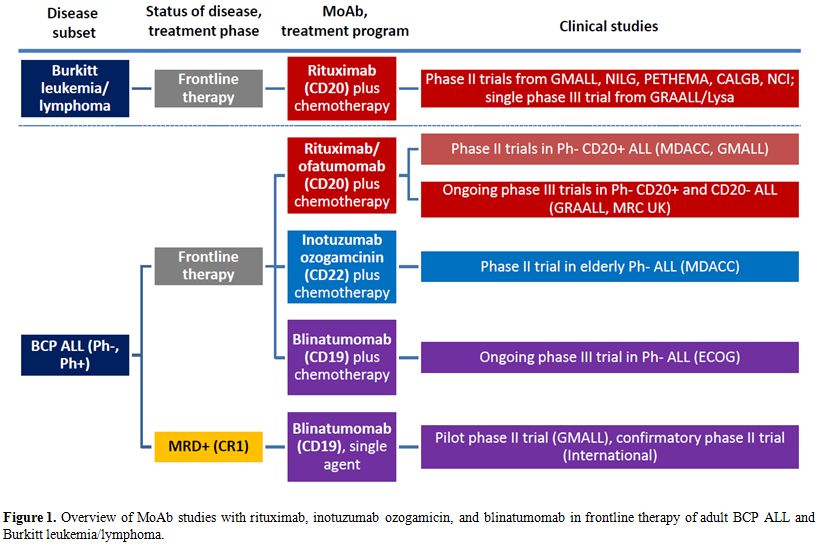 |
Figure 1. Overview of MoAb studies with rituximab, inotuzumab ozogamicin, and blinatumomab in frontline therapy of adult BCP ALL and Burkitt leukemia/lymphoma. |
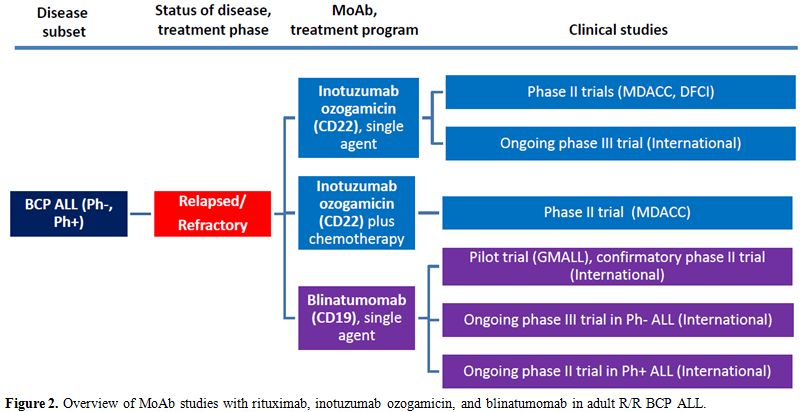 |
Figure 2. Overview of MoAb studies with rituximab, inotuzumab ozogamicin, and blinatumomab in adult R/R BCP ALL. |
Naked Antibodies
Rituximab
and Ofatumomab: anti-CD20 MoAb. The CD20 receptor is the
target of chimeric monoclonal antibody Rituximab. CD20 is expressed by
approximately 40% of BCP ALL cases and virtually any case of mature
B-ALL (Burkitt leukemia). The CD20 receptor functions as a calcium
channel playing a role in cell cycle and differentiation. Rituximab
works as a classical MoAb, reacting at one terminus (Fab/Fv) with the
CD20 epitope on the cell membrane, while the other end (Fc) binds to
complement and Fc receptors of effectors cells. The ensuing MoAb-target
cell interaction activates a complement-mediated cell lysis and/or an
antibody-dependent cellular cytotoxicity (ADCC). Importantly, CD20
expression in CD20 + ALL is upregulated by corticosteroids, which are
commonly given in prephase and continued for several days during
induction therapy.[10,12]
Nothing is known about rituximab activity as a single agent in ALL, and
contrary to other MoAbs experience in R/R ALL is very limited. One
study indicated a response rate of 44% in 9 patients treated with a
rituximab-chemotherapy combination.[13]
Rituximab was instead used in first-line phase II and III programs, and
is used in Burkitt leukemia/lymphoma in adjunct to aggressive
rotational drug regimens.
The usual rituximab schedule in these studies was 375 mg/m2 for
four-eight times, throughout induction and consolidation blocks. A
randomized trial in Burkitt lymphoma confirmed the usefulness of adding
rituximab to intensive chemotherapy blocks, in both HIV negative and
positive HIV patients.[14,15] Several other Burkitt leukemia/lymphoma
regimens reported high response rates, with a curability rate
consistently above 50% and most often between 70-80% and close to
90%-100% in fit patients younger than 55-60 years.[14-21] This means an
average 20% or more improvement over prior results obtained with
similar chemotherapy regimens without rituximab, with no substantial
difference in toxic side effects. Nowadays rituximab is part of the
standard of care for Burkitt leukemia/lymphoma.
About rituximab in frontline therapy of BCP ALL, there were two
randomized trials and two phase II trials in Ph- ALL, all evaluating
its role in addition to induction and consolidation chemotherapy. In
the GRAALL (France/Belgium/Switzerland) phase III trial, CD20+ BCP, ALL
patients (CD20 expression >20%) were randomized with a 2x2
design concurrently testing an augmented cyclophosphamide dose; whereas
in the randomized MRC (United Kingdom) trial, all BCP ALL patients were
randomized to assess the role of the concomitant corticosteroid therapy
in upregulating CD20 expression in CD20- patients. The results from
these two controlled studies are not yet known and are awaited with
interest. As to phase II trials, in the MD Anderson Hospital study[22]
two sequential CD20+ BCP ALL patient cohorts receiving Hyper-CVAD
chemotherapy with or without rituximab were analyzed. In patients aged
60 or less, the CR rate in the rituximab arm was 95% and 3-year
survival 75% (n=68) compared with 47% without rituximab (n=46;
P=0.003), with a proportional increase in MRD negativity evaluated by
flow cytometry (81% vs 58%). A subsequent update showed for the
rituximab-treated group a CR duration of 69% at 3 years with an OS of
71%.[23] In the small group of patients older than 60 (n=16), the CR
rate was high (88%) but the OS was only 29%. The other first-line phase
II trial was from GMALL (Germany) with rituximab added to the 07/2003
chemotherapy schema.[24] This report compared 181 rituximab-treated
patients with 82 pre-rituximab patients. In SR patients (n=196), CR
rate was 94% with rituximab and 91% without; however, minimal residual
disease (MRD) response, evaluated molecularly at week 16 (<10-4)
and, 5-year survival were both improved in the rituximab group, from
59% to 90% and from 57% to 71%, respectively. Similarly, in HR patients
(n=67), CR rate was 81% with rituximab and 88% without; and MRD
response and 5-year survival were improved from 40% to 64% and from 36%
to 55%, respectively. Toxicities were comparable in the two cohorts. In
summary, rituximab could improve the long-term outcome of patients with
CD20+ BCP ALL and seems to enhance the MRD response to induction and
early consideration therapy. This issue arises considerable interest,
given the strict relationship between MRD and outcome in adult ALL and
the dramatically worse outcome of MRD+ CD20+ ALL as opposed to MRD-
CD20+ ALL.[25] Although the CD20 antigen is expressed in a relevant
proportion of Ph+ ALL cases, there is no data on the therapeutic role
of this MoAb in this subset. The most significant data relative to the
use of rituximab in B-lineage ALL Burkitt leukemia/lymphoma are
summarized in Table 1.
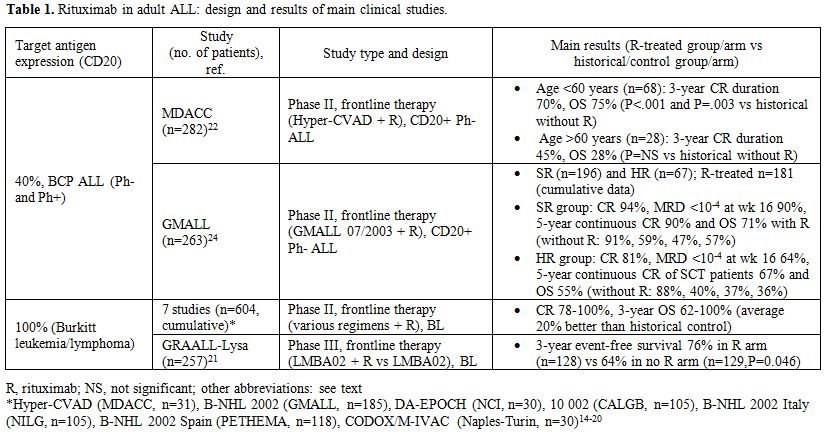 |
Table 1. Rituximab in adult ALL: design and results of main clinical studies. |
Ofatumumab is another anti-CD20 MoAb, which binds to a
different epitope on the CD20 molecule than rituximab, resulting in
greater complement-dependent cytotoxicity. One study evaluated
ofatumumab added to the Hyper-CVAD regimen as frontline therapy of
adult patients with CD20+ ALL.[26] With this regimen, 22 of 23
evaluable patients achieved CR (95%) and were MRD-negative (by flow
cytometry) after cycle 1. One-year remission and OS duration was 91%.
Epratuzumab:
anti-CD22 MoAb. Epratuzumab is a humanized MoAb targeting
CD22. The CD22 antigen is a transmembrane sialoglycoprotein expressed
explicitly by B lymphoid cells. It is expressed on 100% of mature
B-cell ALL and up to 90% of BCP ALL.[27] CD22 regulates B-cell
activation and the interaction of B-cells with T-cells and
antigen-presenting cells. Because of that, CD22 is a good therapeutic
target in BCP ALL. CD22 is rapidly internalized after binding the MoAb
so that the exposure to epratuzumab results in downregulation of B-cell
activation and signaling, with proliferation inhibition.[28] In a phase
I protocol of the Children’s Oncology Group (COG), applied to children
with R/R BCP ALL, 15 children received four doses of epratuzumab twice
weekly for two weeks, then four weekly doses with a standard
reinduction chemotherapy. MRD was evaluated by flow cytometry, and the
absence of MRD was defined as complete molecular remission (CMR). At
the end of the six-week reinduction therapy, nine patients were in CR,
and seven of them were in CMR. Two patients had dose-limiting toxicity,
one grade four seizure, and one grade 3 transaminase elevation. A
subsequent phase II trial (COG ADVL04P2)[28,29] enrolled 114 patients
between 2-30 years of age in first relapse, comparing two different
epratuzumab schedules in addition to traditional reinduction
chemotherapy. The CR rate was comparable in the two study arms
(epratuzumab weekly x 4 doses versus epratuzumab twice weekly x 8
doses: CR 65% vs. 66%) and not significantly higher than the historical
control. The CMR rate was however higher in epratuzumab-treated
patients (42%) than historical controls.
The adult trial SWOG S0910[30] evaluated 32 R/R ALL patients treated
with epratuzumab (4 weekly doses) in association with clofarabine and
cytarabine. The CR rate was 45%, significantly higher than the 17% CR
rate observed in a similar trial with clofarabine/cytarabine without
epratuzumab.[31]
Two other recent reports available only in abstract form concerned a
phase I escalation study of 90yttrium- labeled epratuzumab
tetratexan[32] and epratuzumab added to vincristine/dexamethasone in
R/R ALL.[33] In the first study (n=17), 2 of six patients treated with
a dose of 10 mCi/m2
achieved CR. In the second trial, including 26 elderly patients, four
patients achieved CR, and one a CR with incomplete platelet recovery.
These are promising results obtained in very poor risk patient
populations. Epratuzumab is well tolerated. The most common adverse
events were myelosuppression and mild to moderate infusion reactions
such as fever, nausea, occasionally seizures, and transaminase
elevation.
Alemtuzumab:
anti-CD52 MoAb. Alemtuzumab is a genetically engineered
humanized anti-CD52 MoAb. CD52 is a
glycosylphosphatidylinositol-anchored membrane glycoprotein expressed
by 70-80% of both BCP ALL and T-ALL, making it an attractive
therapeutic target. Alemtuzumab has demonstrated significant activity
in chronic lymphocytic leukemia but was not found effective as a single
agent in acute myeloid leukemia and ALL.
In R/R ALL alemtuzumab was tested in a small adult series of 6 patients
(3 with Ph+ ALL) at the dose of 30 mg given by subcutaneous route three
times weekly for 4-12 weeks (no CR) and was also scarcely effective in
a pediatric trial on 13 patients (one CR).[34,35]
In untreated patients, alemtuzumab was administered as a single agent
in a CALGB trial[36] after three intensive chemotherapy modules in an
attempt to lower post-remission MRD. In 11 evaluable patients, there
was a 1-log median MRD reduction and a noteworthy DFS (median 53
months), but follow-up was provided only for 14 surviving patients. Of
note, the use of alemtuzumab was associated with CMV infection in 8 of
24 patients and herpes virus infection in 5 patients.
For these reasons, alemtuzumab, albeit partially effective, is unlikely
to be developed any further in ALL therapy. It causes a drastic
reduction of lymphocytes including CD4+ and CD8+ T cells predisposing
to opportunistic infections such as CMV and other viruses and
fungi.[36] Thereafter, it requires careful patient monitoring with
serial CMV DNA determinations for pre-emptive therapy, as well as an
adequate anti-infectious prophylaxis.
Immunotoxins and Immunoconjugate Antibodies
Inotuzumab
Ozogamicin: anti-CD22 MoAb. Inotuzumab ozogamicin (IO) is
an anti-CD22 MoAb conjugated to calicheamicin, which is a powerful
anthracycline-like drug. Calicheamicin, a natural product of
Micromonospora echinospora,[37] is a potent cytotoxic agent enabling
cell killing even in the presence of relatively few target sites.
Although CD22 expression is required, IO-related apoptotic effect is
entirely mediated by calicheamicin and not by CD22 signaling. IO is
rapidly internalized and delivers calicheamicin intracellularly. The
toxin binds the minor DNA groove breaking the double-stranded DNA in a
sequence-specific manner.
Forty-nine patients with R/R ALL were treated in a phase I/II trial at
MD Anderson Hospital with single agent IO.[38] Their median age was 36
years and range 6-80 years. All patients had greater that 50% CD22
expression on lymphoblasts, and the majority were heavily pretreated. A
starting dose of 1.3 mg/m2
was used, subsequently increased to 1.8 mg/m2.
The CR rate was 18% and another 39% of the patients had a CR with
incomplete hematologic recovery (CRi), for an overall response rate of
57%. Among the 27 patients who achieved a hematological response, 17
(63%) attained an MRD remission (flow cytometry). Median response
duration was six months, with a trend to improved survival for the 13
patients treated at first salvage. This study, updated including 90
total patients, confirmed the previous results (CR 19%, CRi 39%);
furthermore, the non-hematological toxicity was reduced using the
weekly schedule.[39] Thus, with IO a morphological CR was obtained in
more than 50% of the subjects treated, in association with a complete
MRD response in the majority of these cases. Most responses were short
lived without proceeding to transplantation (n=36), however the
obtaining a CR with associated MRD response, the absence of a complex
karyotype such as t(4;11), t(9;22), or an abnormal chromosome 17 and a
disease status at first salvage were predictive of an improved outcome
with a survival probability of 42+ months.[40] A negative MRD was
observed in 72% of the patients achieving CR/CRi. A new trial for R/R
ALL incorporated IO into a reduced intensity Hyper-CVAD regimen.[41] Of
35 patients treated, 18 (51%) entered CR, 6 (17%) CRi and 1 (3%) marrow
CR, and 12 of them could proceed to allogeneic SCT. Median survival of
responders was 14 months and was not reached in patients at first
salvage. The outcome of IO-treated patients proceeding to allogeneic
SCT was examined separately.[42] The study analyzed the outcome of 26
such patients, of whom 23 were in CR at time of transplant (15
MRD-negative) and three were not. MRD-negative patients had the best
outcome with a 1-year survival of 42%. However, non-relapse mortality
was high in relation to liver toxicity (40% at six months), with 5
deaths by venoocclusive disease. These results could improve choosing
the less hepatotoxic conditioning regimens and concomitant drugs. In
conclusion, these single-center studies IO brought more patients with
R/R ALL to allotransplantation (45%) than chemotherapy, but the salvage
rate was affected by transplant-related toxicity, indicating the need
of a careful design of all treatment components. An international phase
III study comparing IO with standard reinduction therapy in R/R ALL is
near to a conclusion.
In untreated patients, IO was added to mini-hyper-CVAD (dose reductions
and no anthracycline) in elderly ALL.[43] Twenty-seven patients aged
60-79 years (median 69 years) were treated, and 25 (96%) entered CR,
all with negative flow cytometry MRD. The 1-year survival was 81%,
superior to the historical control group. Although the follow-up is
short, these are outstanding induction results obtained in a high-risk
patient population. Another US Intergroup trial is planned in patients
aged 18-39 years, adding IO to the C10403 chemotherapy backbone.
On the toxicity side, IO is myelotoxic, as reflected by the high rates
of CRi. Grade 3-4 non-hematologic adverse events included drug-related
fever (18%) with hypotension, hyperbilirubinemia (4%) and transaminase
elevation (1%). All the events but the increased bilirubin were
reversible. A biopsy demonstrated liver fibrosis in two patients. A
venoocclusive disease of the liver was reported in 5/22 patients after
allogeneic SCT.[39] However, 4 of 5 of these patients received a
preparative regimen of clofarabine/thiotepa. Furthermore, two distinct
reports suggest a benefit toward liver toxicity with weekly rather than
single dose IO administration.[39,44]
The most significant data relative to the use of IO in B-lineage ALL
are summarized in Table 2.
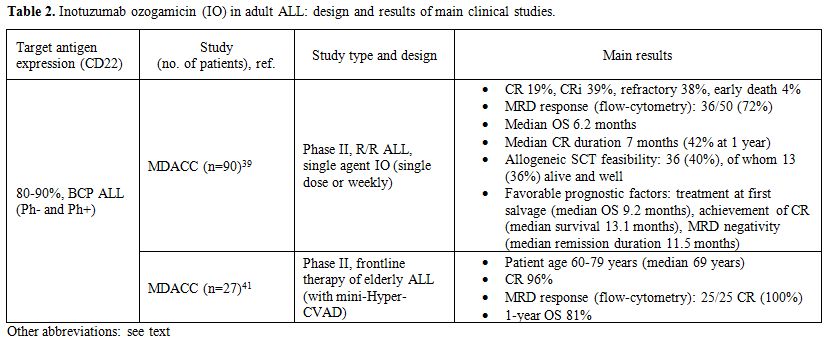 |
Table 2. Inotuzumab ozogamicin (IO) in adult ALL: design and results of main clinical studies. |
BL22
and CAT-8015: anti-CD22 MoAbs. Because the CD22
antigen-immunotoxins is rapidly internalized, CD22 is an attractive
therapeutic target.[45] The first-generation immunotoxins BL22
demonstrated cytotoxicity in vitro and also in vivo and in a phase 1
trial. A decrease of leukemia blasts was observed in 16 out 23 ALL
patients, but no CR was obtained.[46] Three of these patients developed
neutralizing antibodies,[47] but no allergic reaction, vascular leak or
hemolytic uremic syndrome occurred.
A second-generation immunotoxins, CAT-8015, was subsequently
developed,[45] trying to reduce non-specific toxicities, increase MoAb
stability and improve activity.[46] In one small trial, 4 out of 9
treated patients achieved a CR.[10] Another phase I trial showed a CR
in 4 out of 19 heavily pretreated children and young adults, plus one
partial response and 8 hematological improvements.[47] Resistance due
to low levels of DPH4 mRNA and target protein was described.[48]
Further analysis of the DPH4 gene promoter demonstrated
hypermethylation in the resistant cells. This mechanism could be
reversed by hypomethylating agents such as 5-azacytidine.
Combotox:
dual anti-CD19/CD22 MoAb. Combotox is a combination of
anti-CD19 and anti-CD22 deglycosylated ricin-A chain immunotoxin.[49]
This treatment has the advantage of targeting two different antigens.
In a pediatric trial, 3 of 17 R/R patients achieved a CR.[50] The
dose-limiting toxicity was a vascular leak syndrome, caused by an
endothelial damage due to a unique aminoacid motif in the ricin-A
toxin. Preclinical studies in murine ALL model demonstrated synergy
with the sequential administration of combotox with cytarabine.
SAR3419
and anti-B4-blocked ricin: anti-CD19 MoAb. SAR3419 is an
anti-CD19 humanized MoAb linked to a highly powerful tubulin inhibitor,
maytansinoid DM4, eliciting ADCC.[51] SAR3419 is internalized and then
routed to lysosomes, whereupon it is degraded to yield the active drug.
In preclinical models, an extended duration of remission was documented
when SAR3419 was administered after an induction regimen as maintenance
therapy.[52] A Phase II trial on R/R ALL is ongoing. Reversible corneal
toxicity was described as dose-limiting toxicity.
The anti-B4-blocked ricin MoAb was used in a frontline CALGB study in
patients with CD19+ ALL instead of high-dose cytarabine consolidation,
reserved to CD19-negative ALL patients.[53] Forty-six patients were
treated. Although feasible, this treatment did not result into an
improved outcome and/or MRD response compared to the other
patients.
Bispecific T-Cell Engager (BiTE®) Antibodies
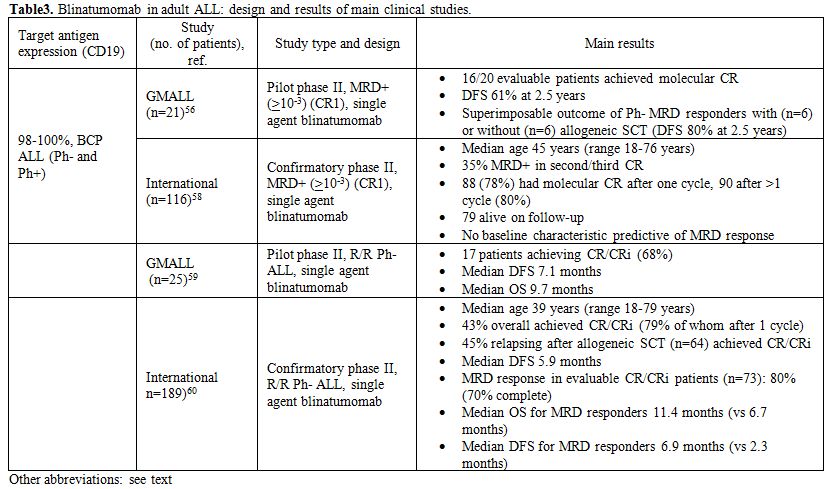 |
Table 3. Blinatumomab in adult ALL: design and results of main clinical studies. |
Modern Immunotherapy with CAR T Cells
A breakthrough in cellular therapy for BCP ALL. Normal autologous or allogeneic T cells can be harvested from patients or normal donors to be genetically modified to express a chimeric antigen receptor (CAR) recognizing specific targets on leukemic cells, then expanded and reinfused in the patient to exert antileukemic activity. A CAR consists of a single chain variable antibody fragment highly specific to a tumor antigen, which is fused to the transmembrane domain and a T cell signaling moiety.[63] The resulting receptor, when expressed on the surface of a T cell, mediates binding of the target tumor antigen and activates a signal to the T cell, inducing target cell lysis. Second and third generation CAR T cells present a single-chain variable fragment that resides outside of the T cell membrane and is linked to stimulatory molecules inside the T cell. The general schema for production of CAR T cells and their in vivo activity against CD19+ ALL cells is shown in Figure 3.
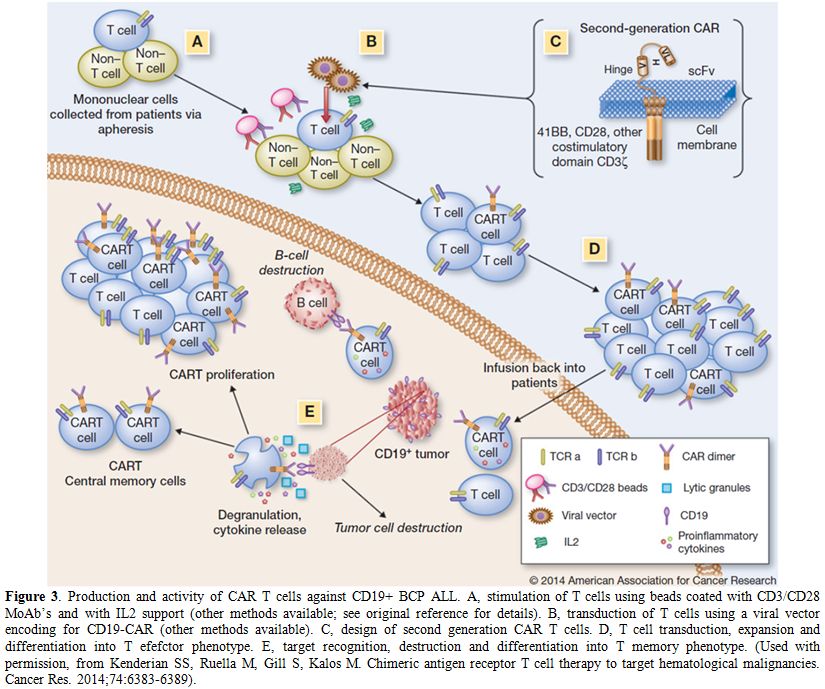 |
Figure 3. Production and activity of CAR T cells against CD19+ BCP ALL. A, stimulation of T cells using beads coated with CD3/CD28 MoAb’s and with IL2 support (other methods available; see original reference for details). B, transduction of T cells using a viral vector encoding for CD19-CAR (other methods available). C, design of second generation CAR T cells. D, T cell transduction, expansion and differentiation into T efefctor phenotype. E, target recognition, destruction and differentiation into T memory phenotype. (Used with permission, from Kenderian SS, Ruella M, Gill S, Kalos M. Chimeric antigen receptor T cell therapy to target hematological malignancies. Cancer Res. 2014;74:6383-6389). |
Clinical studies with CAR T cells. CAR T cells with specificity for CD19 have shown promising results in chronic lymphocytic leukemia.[64] Preliminary results of this approach used in two children with R/R ALL were published.[65]
In one case there was a sustained remission. Other recent pre-clinical
studies support additional genetic modifications to achieve optimal
clinical efficacy.[66-68] Altogether, there is
accumulating evidence pointing to the relevant activity of CD19-CAR T
cells and CD22-CAR T cells in R/R ALL. These patients are usually
prepared with immune suppressive therapy before receiving the CAR T
cell infusion (with cyclophosphamide and fludarabine). A breakthrough
publication[69] demonstrated the potential of this
treatment in 5 adult patients with R/R ALL (age range 23-66 years). At
time of CAR T cell therapy, 3 patients were refractory to salvage
chemotherapy, and one was MRD+. After CAR T, all were in clinical and
molecular CR, and 4 out of the 5 patients could undergo an allogeneic
SCT. Other reports soon followed with either CD19-CAR T or CD22-CAR T,
expanding our knowledge about this innovative treatment method.[70-73]
Two very recent publications reported the final results of prospective
trials using CAR T cells obtained through different methodology on 30
and 21 patients with relapsed ALL, respectively, including a few adult
subjects.[74,75] In the first study autologous
CD19-CAR T cells induced a CR in 27 (90%) patients (of whom 2 had
previously failed blinatumomab, and 15 had relapsed following
allogeneic SCT). The event-free survival was 67% at 6 months,
associated with persistence of CAR T cells (68%) and B-cell aplasia
(73%). In the second trial, aimed at establishing the maximum tolerated
dose of CAR T cells (defined as 1x106/kg
CAR T cells), the generation of CAR T cells was successful in 20 of 21
patients (90%). Treatment toxicity mediated by cytokine release was
fully reversible, prolonged B-aplasia did not occur, and 14 patients
got a CR (70%) including 6 of 6 with primary refractory ALL. Moreover,
12 patients achieved MRD negative status, and 10 proceeded to
allogeneic SCT. In both studies CAR T cells were detectable in the
cerebrospinal fluid, clearing off blast cells in some patients with
meningeal leukemia.
Presently, CAR T cell treatment remains
experimental and available only at selected centers due to its
technical complexity. It is however highly promising and must be
developed further as a potential major step forward in the management
of adult BCP ALL. CAR T cells carry peculiar toxicities related to cell
expansion/activation, resulting in a cytokine- release syndrome which
is occasionally associated with cardiorespiratory failure requiring
admission to intensive care unit. The interleukin-6 inhibiting agent
tocilizumab is effective in this setting. The degree to which this
treatment causes a permanent B-cell depletion with severe
hypogammaglobulinemia in long-term survivors is another critical point.
Conclusions
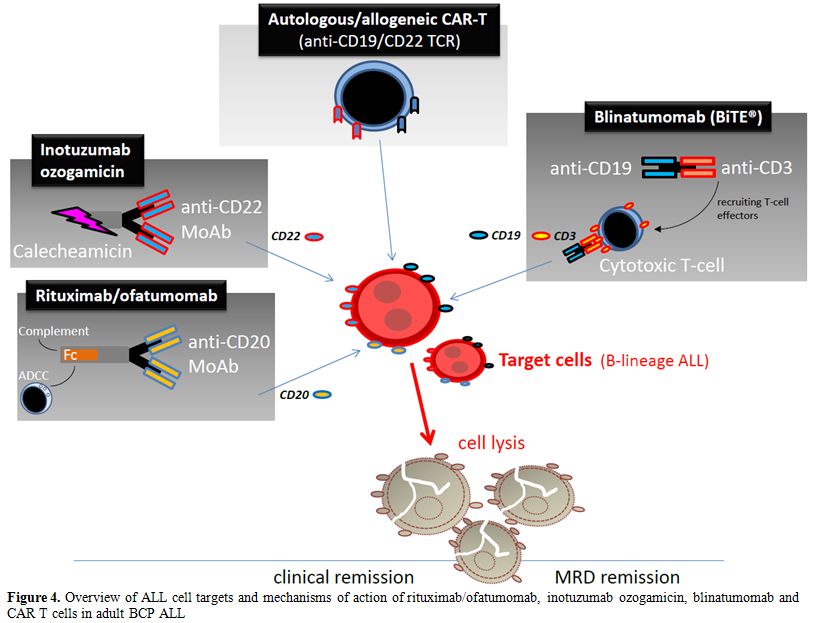 |
Figure 4. Overview of ALL cell targets and mechanisms of action of rituximab/ofatumomab, inotuzumab ozogamicin, blinatumomab and CAR T cells in adult BCP ALL. |
References

[TOP]