Received: August 20, 2014
Accepted: November 24, 2014
Mediterr J Hematol Infect Dis 2015, 7(1): e2015004, DOI 10.4084/MJHID.2015.004
This article is available on PDF format at:

Monica Sharma, Sanjay Pandey, Ravi Ranjan, Tulika Seth and Renu Saxena
Department of Hematology, AIIMS, New Delhi
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Introduction. Cases with microcytosis not responding adequately to iron supplementation are diagnostic dilemma and have been reported to harbor alpha (α) thalassemia mutations. The aim of this study was to determine the common α globin gene deletions in cases with microcytic anemia.Methods Fifty four patients selected (22 females and 32 males) had microcytic anemia (MCV < 80 fl, Hb <12gm/dl) with raised TRBC (> 5M/mm3) but normal Hb HPLC. They had either low or normal Transferrin Saturation (TS). Gap-PCR for four common α-gene deletions (-α3.7, -α4.2, - -αSA and --αSEA) was done. Results Out of the total fifty-four cases nineteen (35.2%) cases were found to have α gene mutations; Three homozygous and sixteen heterozygous cases including -α3.7 deletions and a single case of --αSA ; but no -α4.2 and –SEA mutations were found. Conclusion α gene mutations can confound iron deficiency anemia, but no RBC indices, or a discriminant function can identify it is presence Molecular studies have to be resorted to. Gap PCR for common α thalassemia mutation including –αSA should be done even in the face of low iron stores in subjects who respond incompletely to iron supplementation. |
Introduction
Iron deficiency anemia (IDA), β thalassemia trait (βTT) and anemia
of chronic disease (ACD) are common causes of microcytosis that can be
diagnosed accurately by Iron studies and Hb HPLC respectively. However,
when HbA2 level is normal or low along with normal or low serum iron
studies, microcytosis can be a diagnostic dilemma. Walford et al. and
Pearson et al.[1,2] had long back suggested that such cases could be
harboring α thalassemia mutation.
The α -thalassemia syndromes
are among the most common single-gene disorders with more than 20% of
the world population to be a carrier of some form of α –thalassemia, as
estimated by The World Health Organization (WHO).[3-5] In India also it
is common, and the gene frequency of occurrence is appreciably higher
than that of βTT.[6] Deletion - α3.7 is the commonest reported
genotype which exists in α + (-α/αα) milder form.[7,8] Most alpha
thalassemia cases in India have been reported among tribal
populations[8,9] with βTT and other hemoglobinopathies but its
co-existence with IDA has never been studied.[7,10-13] Iron deficiency
is highly prevalent in India and with the reportedly high frequency of
α thalassemia the likelihood of the two conditions coexisting can be
expected to be high. The Discrimination between microcytosis due to the
two conditions is not only clinically significant but is often
difficult even more so when they coexist. No Discriminant function or
RBC Indices can indicate the presence of α thalassemia in the subjects
with or without IDA.[14-16] Molecular studies most commonly
deletion-specific gap-PCR have to be resorted to for detection of the
common α0- and α-thalassemia
deletions in such cases when the Hemoglobin or microcytosis does not
improve appropriately after iron supplementation.
The mutation
spectrum for each population group is distinct characterized by a small
number of founder mutations that reflect the predominant mutant alleles
in carriers and affected individuals.[17,18] This helps in designing
the gap PCR assay individually or in multiplex panels. One of the most
popular multiplex assays covers seven deletions, specifically the SEA,
FIL, MED-I, THAI, 20.5, 3.7, and 4.2 deletions.[19-22] Other molecular
studies though not readily available are multiplex ligation-dependent
probe amplification (MLPA) /microarray analysis, allele-specific assays
and α -globin gene resequencing for detection of common, rare, and
private point mutations.
Based on previous studies in India in our laboratory, gap PCR for - α3.7, -α4.2,- - αSA and - - αSEA deletions are done.
Microcytic
hypochromic anemia is the hallmark feature of α thalassemia, and the
degree of microcytosis is directly proportional to the number of alpha
genes deleted, thus, reflecting the rate of imbalance between α - and
β-chain expression.[23,24] Apart from few studies,[15,25] there is a
paucity of population-based studies on MCV in α thalassemia observing
the effect of its variation in alpha genes deletions & also it is
an alteration with the coexistent IDA.
This study was undertaken
to identify and highlight the presence of common α thalassemia
deletions in cases of microcytic anemia and its interaction with IDA.
Materials and methods
Patients:
Fifty-four Patients, attending Hematology outpatient department, All
India Institute of Medical Sciences, New Delhi were included in the
study that was approved by institutional ethical committee. Duration of
the study was 1.5 years between 2010 and 2011.The cases included had
Hemoglobin <12 gm/dl, MCV<80fl and TRBC > 4.5 M/mm3. Only
cases with standard Hb HPLC showing low or normal HbA2 (< 3.5 %)
were taken to exclude βTT/Hemoglobinopathy.
Sampling:
3 ml blood in EDTA for CBC and Hb HPLC and 1 ml blood in 3.2% sodium
citrate for DNA studies was obtained after taking signed consent from
the patients.
Hematological work up: Hematological parameters were
analyzed by automated cell counter (XT1800i, Sysmex Transasia).
Quantification of HbA2 was performed by High Performance Liquid
Chromatography (Hb-HPLC, Bio-Rad variant- β thalassemia short program).
Serum Iron studies were performed by standard laboratory methods and
transferrin saturation (TS) <16% was taken to be iron-deficient.
Detection of the α thalassemia mutations:
Genomic DNA was prepared from peripheral blood by the standard
phenol-chloroform extraction method. Deletion mutations were
characterized by Gap-PCR. Detection for single deletion - α3.7 and -α4.2 Baysal et al.[26] - - αSA deletion Shahji et al.,[5] and - - αSEA Chang et al. was done.[27]
Follow-up:
Though it was intended to be a cross-sectional study, the Follow-up
data on Iron therapy available were evaluated in 27 (54%) cases.
Statistical analysis:
The following parameters were assessed: Hemoglobin, MCV, TRBC, RDW-CV,
TS and HbA2. Mutation positive and negative groups were compared.
Statistical analysis was carried out using the statistical package SPSS
version,[15] and an independent sample t-test was used for comparison
of hematological parameters. Statistical significance was assessed as a
test with p ≤ 0.05.The hematological parameters of three alpha
thalassemia mutations, the four groups based on mutation status and TS
< / > 16 % were also compared, but the size in each group was too
small for obtaining a valid statistical comparison.
Results
Patient characteristics:
Of the 54 cases included in the study, 32 (57.4%) were males and 22
(42.6%) females and the median age was 19.5 years (13 months - 62
years). There were four pediatric cases (<18 years) in alpha
positive genotype group and seven in wild alpha genotype group. The
subjects were, for the most, anemic (median Hemoglobin was 9.6 g/dl
(5.1-12g/dl), with RBC showing microcytosis (median MCV63.4fl (48-79
fl) and iron deficiency (median TS 10.2% (7.6-19.2%).They had TRBC
median 5.2M/mm3 (4.5-6.2M/mm3), median RDWCV 19.3 %( 14-29%) and median HbA2 2.3 %( 1.6-2.8%).
Characteristics of alpha thalassemia subjects:
Nineteen out of 54 (35.2 %) cases showed α thalassemia deletions and
three genotypes were seen on gel electrophoresis. Fifteen were –α3.7
heterozygous, three were α3.7 homozygous and one was - - αSA double deletion (Figure 1 and 2).
Comparative hematological parameters of mutation-positive and
mutation-negative cases are given in table 1. The subjects had
presented chiefly for work up for long standing anemia or detected
incidentally and few for nonimmune hydrops. The median hemoglobin was
10.5 g/dl (5.9-11.9g/dl) across the α thalassemia genotypes. The median
MCV was significantly lower in the three homozygous with α3.7 deletions, median 56.2fl (56-72.4fl), compared to the median, 68.9fl (47.6-80fl) (29.6%), of the subjects -a3.7 deletion heterozygous and to the value, 71.6fl, of the one subject (1.9%) –with αSA thalassemia.
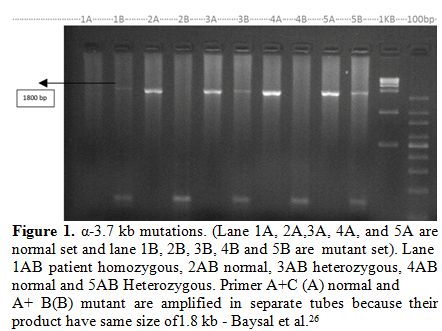 |
Figure
1. α-3.7 kb mutations. (Lane 1A, 2A,3A, 4A, and 5A are normal set and
lane 1B, 2B, 3B, 4B and 5B are mutant set). Lane 1AB patient
homozygous, 2AB normal, 3AB heterozygous, 4AB normal and 5AB
Heterozygous. Primer A+C (A) normal and A+ B(B) mutant are amplified in
separate tubes because their product have same size of1.8 kb - Baysal
et al.[26] |
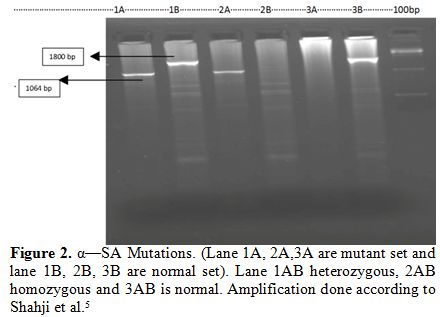 |
Figure 2. α—SA Mutations. (Lane 1A, 2A,3A are mutant set and lane 1B, 2B, 3B are normal set). Lane 1AB heterozygous, 2AB homozygous and 3AB is normal. Amplification done according to Shahji et al.[5] |
There were high RDW-CV and low %TS indicating coexisting IDA
in 10 of the 19 mutation-positive cases. On comparing the hematological
parameters of α thalassemia mutation-positive and negative cases (Table 1).
The mean Hemoglobin and MCV were better in the mutation-positive cases
while the mean RDW-CV was higher in the mutation-negative group
(Hemoglobin, MCV p=0.161, p=0.151, RDW-CV and TS p=0.040, 0.001
respectively). Interestingly 28/35(80%) mutation negative cases showed
raised TRBC (Table 2) suggesting that a high >5M/mm3 TRBC may not always be thalassemia.
Effect of coexisting iron deficiency on α thalassemia RBC indices (Table 3):
The median Hemoglobin, MCV, and HbA2 were lower in both the iron
deficient groups 1 and three, but the values were relatively much lower
in the mutation-negative group 1which was predominantly IDA. TRBC was
high in both the groups. Between groups 2 & 4 which were both iron
sufficient (i.e. TS > 16%) the median Hemoglobin, MCV, TRBC, and
HbA2 were lower in group 4 (mutation positive) compared to group2. The
RDW-CV was comparable indicating the coexisting IDA in both the groups.
This shows that MCV is lower in α thalassemia compared to Normal.
Follow up on Iron was available in 27 (54.00%) cases. Response was seen
predominantly (63.1%) in group 1 as expected but 36.8% in this group
also failed to respond no details about the compliance or cause of
refractoriness was available. Subjects of Group 2 did not require iron,
but they needed to be investigated for other underlying
Hemoglobinopathy, which was beyond the scope of this study. In
mutation-positive group 3 (50%) patients showed no or inadequate
response to iron therapy while individuals in group 4 had % TS >16%
and did not need iron supplementation. The family history too was
significant only in individuals of the group 4.
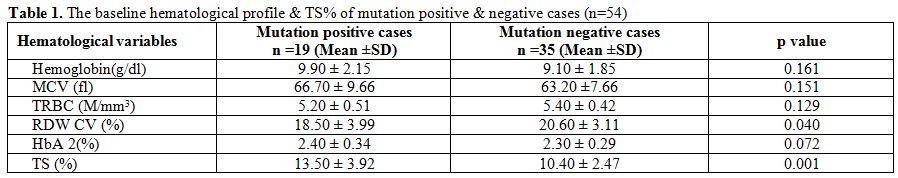 |
Table 1. The baseline hematological profile & TS% of mutation positive & negative cases (n=54) |
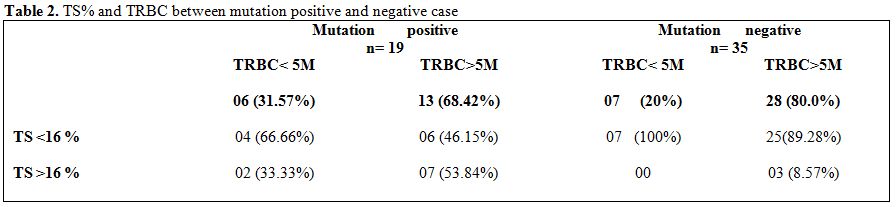 |
Table 2. TS% and TRBC between mutation positive and negative case |
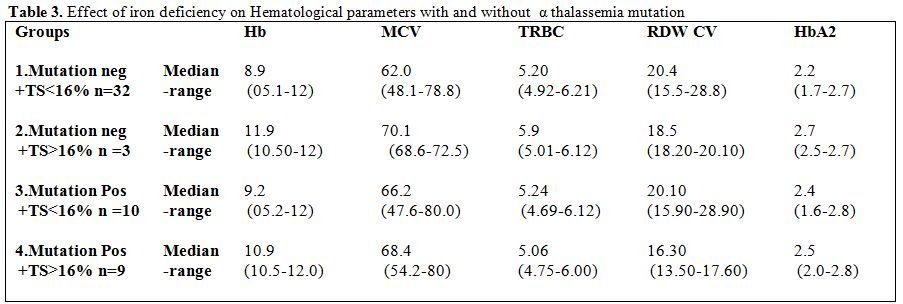 |
Table 3. Effect of iron deficiency on Hematological parameters with and without α thalassemia mutation |
Discussion
Fifty-four subjects with microcytosis but with normal Hb HPLC and
normal or low Iron studies were evaluated for the presence of common α
-thalassemia deletion mutations. Microcytosis was the defining criteria
in the study, but it is also important to remember that normal RBC
indices do not rule out α thalassemia carrier.[28] In the current study
with gap, PCR α thalassemia could be detected in nearly 1/3rd
(35.2% -19/54 cases) of the microcytic cases. These results agree with
50% a-thalassemia cases reported in non-anemic microcytosis cases by
Borges et al.[29] in southeastern Brazilian population and the other
studies in European or European-derived populations who also have
reported a-thalassemia trait in 25%-80% of non-anemic subjects with
microcytosis without iron deficiency.[30,31] As reported in various
Indian studies -α3.7 deletion was the
commonest determinant in the study seen predominantly in the
heterozygous state (31.7% of microcytic patients).[7,11] Interestingly
a single case of --αSA was detected incidentally.[5] Though 5% and 3.33% cases of -α4.2 , --αSEA
deletion respectively have been reported in Indian subjects by Sarkar
et al.[32] but none was seen in the study which could have been due to
the difference in the ethnic group which in this study was largely
North Indian. This also could explain the high frequency of
a-thalassemia cases observed even when only microcytic subjects were
investigated, and uncommon α thalassemia deletions and point mutations
could not be done. This is concurrent with the observations of K Ghosh
et al who have reported highest prevalence in the Punjabis.[7]
Phenotypically
all the 19 α thalassemia cases were very similar presenting with only
mild anemia. None of the cases had jaundice or gall stone disease.
Family history was positive in four individuals. Microcytosis was the
defining criteria and was most pronounced in the three (5.6%) -α3.7 deletion homozygous individuals compared to (29.6%) -α3.7 deletion heterozygous and one (1.9%) --αSA
thalassemia subjects. Though insufficient in number, the findings are
largely in concordance with previous reports, where microcytosis has
been explained on the basis of α-gene number.[29,32-36] IDA causes the
Indices to be much lower than in α thalassemia and may have high TRBC
in pediatric group and after recent iron supplementation.[1,2,37,38] A
report on alpha thalassemia with anemia in children’s revealed it
should be considered differential diagnosis.[39] On comparing the
hematological parameters within the four groups, group 1( IDA cases)
had the most pronounced microcytosis even more than the cases with IDA
coexistent with α thalassemia mutation (group 3) which had more than
that of α thalassemia cases (group 4).[1] (MCV group1 <group3<
group4 < group2). The subjects of Group 3 also had much lower RBC
indices than that of group 4 which was akin to normal individuals. In
conclusion, Using low Hemoglobin & MCV ,raised TRBC, normal Hb A2
and deletion specific gap- PCR for common alpha thalassemia mutations
(including - -αSA), nearly 1/3
(35.2%) of the 54 cases of microcytic anemia with or without IDA could
be typed in the study. Finding of IDA should not be a deterrent for the
screening of a coexisting α thalassemia mutation, which is particularly
relevant in a country like India where both α thalassemia (in some
geographical areas) and IDA occur with high frequency. Larger
population studies, including other less common α thalassemia
mutations, are required not only to identify the population group
specific mutation spectrum but also to observe the interplaying of the
two conditions on the RBC indices. In fact red cell indices could
differentiate or indicate the coexistence of the two conditions. Until
then molecular studies (gap-PCR) should be done for the detection of
common α thalassemia mutations especially when the Hemoglobin or
microcytosis does not improve appropriately on iron supplementation
this can be useful in the countries where α thalassemia is not
prevalent because as a consequence of population migrations alpha
thalassemia has acquired a truly global distribution.
References

[TOP]