Received: March 13, 2015
Accepted: April 20, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015034, DOI 10.4084/MJHID.2015.034
This article is available on PDF format at:

Ulrike Bacher, Julie Schanz, Friederike Braulke and Detlef Haase
Department of Hematology and Medical Oncology, University Medical Center Göttingen, Göttingen, Germany
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract The karyotype represents one of the
main cornerstones for the International Prognostic Scoring System
(IPSS) and the revised IPSS-R (IPSS-R) that are most widely used for
prognostication in patients with myelodysplastic syndromes (MDS). The
most frequent cytogenetic abnormalities in MDS, i.e. del(5q),
-7/del(7q), +8, complex karyotypes, or –Y have been extensively
explored for their prognostic impact. The IPSS-R also considers some
less frequent abnormalities such as del(11q), isochromosome 17, +19, or
3q abnormalities. However, more than 600 different cytogenetic
categories had been identified in a previous MDS study. This review
aims to focus interest on selected rare cytogenetic abnormalities in
patients with MDS. Examples are numerical gains of the chromosomes 11
(indicating rapid progression), of chromosome 14 or 14q (prognostically
intermediate to favorable), -X (in females, with an intermediate
prognosis), or numerical abnormalities of chromosome 21. Structural
abnormalities are also considered, e.g. del(13q) that is associated
with bone marrow failure syndromes and favorable response to
immunosuppressive therapy. These and other rare cytogenetic
abnormalities should be integrated into existing prognostication
systems such as the IPSS-R. However, due to the very low number of
cases, this is clearly dependent on international collaboration.
Hopefully, this article will help to inaugurate this process. |
Introduction
Myelodysplastic Syndromes (MDS) representing clonal hematopoietic
stem cell disorders were shown to be highly heterogeneous from
clinical, phenotypic, cytogenetic,[1,2] and, more recently, from molecular genetic aspects.[3-5]
Cytogenetic abnormalities are detectable in 40-60% of patients with de
novo MDS and in up to 90% of patients with therapy-associated MDS
(t-MDS) or secondary MDS.[6] Together with the bone
marrow blast percentage and peripheral blood values (hemoglobin,
neutrophils, thrombocytes), the karyotype of the hematopoietic cells
provides the basis for the IPSS-R (international prognostic scoring
system)[2,7] and is essential for
therapeutic decision making in patients with this heterogeneous
disorder. This is of utmost importance considering the range of
possible therapeutic strategies[8] including purely
supportive concepts, treatment with drugs including lenalidomide and
demethylating agents (azacitidine and decitabine), or allogeneic
hematopoietic stem cell transplantation (HSCT) for high-risk MDS
patients.[9] The karyotype furthermore has an
important role establishing the diagnosis of MDS. During the course of
the disease, karyotyping contributes to assess the response to therapy
or may identify clonal evolution as a sign of progression.[10]
The
most frequent cytogenetic abnormalities in MDS such as del(5q),
-7/del(7q), +8, complex karyotypes, or –Y have been extensively
explored for their prognostic impact. Some less frequent isolated
cytogenetic abnormalities have been characterized for their impact on
prognosis such as del(11q) (very good prognosis), isochromosome 17q or
+19 (intermediate prognosis), or inv3/t(3q)/del(3q) (poor prognosis).[2] These results found already entrance in the IPSS-R.[7] However, there is a much larger variety of cytogenetic abnormalities in MDS. In a previous study[1]
investigating a large cohort of more than 2,000 patients with MDS, a
total of 684 different cytogenetic categories were identified. The wide
spectrum of less frequent cytogenetic categories includes e.g. gains of
chromosomes 1 or 1q, 14 or 14q, gains or losses of chromosome 21, or
loss of one X-chromosome.[1] Many of those rare
cytogenetic abnormalities are associated with distinct prognostic
profiles. So far, these rare cytogenetic abnormalities only found
limited attention in MDS studies. Aiming to focus interest on this
issue, this review article discusses the role of several selected rare
cytogenetic abnormalities (Table 1) in patients with MDS. Cytogenetic aberrations that had already been considered within the IPSS-R[2,7]
(i.e. cytogenetic abnormalities that were detected in more than 10
patients in the large cohort of patients with MDS were representing the
basis for the cytogenetic scoring system of the IPSS-R)[2]
were not included in this review article. As a complete summary of all
relevant rare abnormalities in MDS was beyond the scope of this
article, the authors focused on a selection of abnormalities that were
considered to be most interesting due to their prognostic relevance,
due to specific biologic characteristics, or due to associations with
specific molecular mutations.
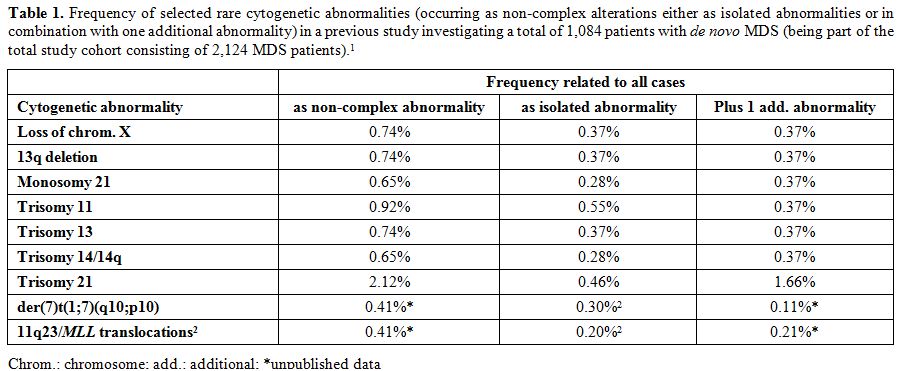 |
Table 1. Frequency of selected rare cytogenetic abnormalities (occurring as non-complex alterations either as isolated abnormalities or in combination with one additional abnormality) in a previous study investigating a total of 1,084 patients with de novo MDS (being part of the total study cohort consisting of 2,124 MDS patients).[1] |
In general, most frequent in MDS are cytogenetic losses
resulting from monosomies or deletions, mostly involving chromosomes 5,
7, 20 or Y. The gain of genetic material with the occurrence of total
or partial trisomies (e.g. of chromosomes 1, 8, 11, and 21) is less
frequent. Unbalanced translocations may also cause losses or gains of
genetic material. Different from AML balanced structural abnormalities
occur only rarely in MDS. Some of the most important rare cytogenetic
abnormalities in MDS are discussed in detail in the following, focusing
on their frequency, their diagnostic meaning, their molecular
background or associations and their impact on the prognosis according
to the literature and own findings.
Chromosomal Losses
Loss of the X-chromosome. Acquired non-constitutional loss of a sex
chromosome (Y-loss in males, X-loss in females) is an age-related
phenomenon, but can also occur in association with hematological
malignancies. [11-14] Usually, aberrations of the sex
chromosomes are numerical losses or gains affecting the whole
chromosome. Structural changes such as partial deletion of X are very
rarely detected as part of unbalanced translocations or very complex
aberrations. The loss of the Y-chromosome as a sole abnormality belongs
to the more frequent cytogenetic abnormalities in MDS and is associated
with a very good prognosis.[2] In contrast, an
acquired loss of the X-chromosome in females is a rare abnormality in
MDS and AML and is described to occur as a single abnormality in 0.2 –
0.3% of patients with these myeloid malignancies only[2,15,16] and in up to 1.5% in combination with other abnormalities.[17]
As a sole aberration, loss of X-chromosome in MDS is associated with an
intermediate prognosis, a median overall survival (OS) of 16 months and
a median leukemia-free survival of 14 months.[2] As a constitutional abnormality, a monosomy X defines Turner´s syndrome,[18,19] but the respective syndrome does not seem to be associated with an increased risk of hematological malignancies.[20-22]
By
FISH a suitable probe for the detection of the sex chromosomes is
easily available and can be performed on different samples (bone
marrow, CD34+ myeloid peripheral blood cells, CD3+ peripheral T-cells).[13,23,24] Further cytogenetic analyses of e.g. PHA- stimulated lymphocytes are needed[11] to distinguish a constitutional monosomy X from an acquired loss of the X-chromosome restricted to the hematopoietic cells.
13q Deletion.
Deletion of 13q occurs in different hematological malignancies, i.e. in
lymphatic malignancies such as chronic lymphocytic leukemia (CLL) or
multiple myeloma, but may also be observed in myeloid malignancies such
as chronic myeloid leukemia (CML), myeloproliferative neoplasms (MPNs),
or MDS (Figure 1). The RB1 gene that had been first identified in patients with retinoblastoma[25] was shown to be located within the common deleted region in leukemic cells carrying a del(13q).[26] The RB1 gene is a tumor suppressor gene that is involved in cell-cycle control and in the process of cell differentiation.[27] FISH with probes for RB1 is a useful adjunct to cytogenetics when abnormalities of 13q14 are detected.[28] In MDS, the frequency of 13q deletion was given around 2%.[27]
In patients with myeloid malignancies and occurrence of 13q deletion, a
strong association to therapy-associated MDS or AML has been described.[27]
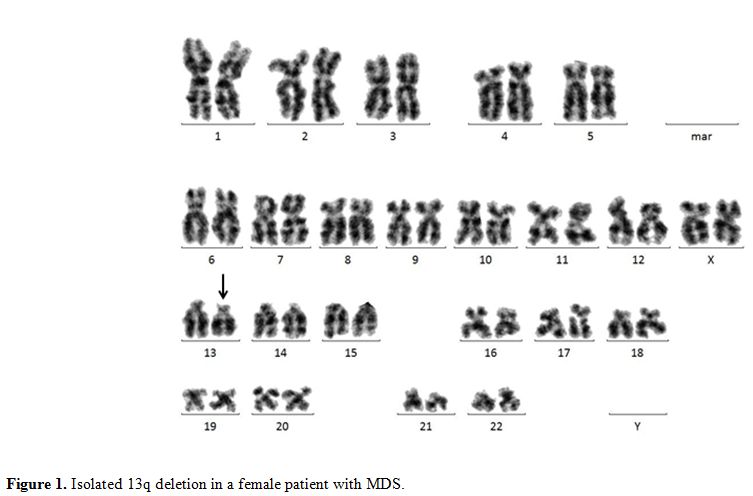 |
Figure 1. Isolated 13q deletion in a female patient with MDS. |
It has been suggested that unclassified MDS (MDS-U) with
del(13q) was a benign bone marrow failure subset characterized by good
response to immunosuppressive therapy and a high prevalence of PNH
clones.[29-31] Hosokawa et al. investigated 22
patients showing bone marrow failure with a sole del(13q) or with a
del(13q) and additional cytogenetic abnormalities. All del(13q)
patients were diagnosed with MDS-U. PNH clones were detected in 19
patients. Strikingly, all 14 patients with del(13q) alone and 2 of 5
patients with additional cytogenetic abnormalities responded well to
immunosuppressive therapy with favorable 10-year OS rates of 83% and
67%. Only two patients with additional cytogenetic abnormalities
developed s-AML.[29] To further investigate this
phenomenon, Holbro et al. analyzed 86 patients with aplastic anemia
(AA) with available cytogenetic results. Six patients (7%) showed a
del(13q). All but one of these patients showed evidence of PNH clones.
Also in this series, there was a favorable response to
immunosuppressive therapy and all patients but one were still alive
after a median follow-up of 105 months. None of the patients with
del(13q) developed leukemic transformation. Thus, the patients with
bone marrow failure and del(13q) from both cohorts (Holbro et al.;
Hosokawa et al.) showed similar characteristics and outcomes. These
reports suggest that MDS-U with del(13q) may be seen as an overlap
condition between MDS and AA.
Monosomy 21. Monosomy 21 (-21) as a sole abnormality is rare in primary MDS with a frequency of 0.3%.[2]
Monosomy 21 may also occur in combination with one more cytogenetic
aberration, reaching a frequency of 0.5% in a large series of patients
with primary and secondary MDS.[1] The frequency of
-21 as part of a complex karyotype is not well defined in the
literature, possibly due to the small number of cases. The knowledge
regarding the prognostic impact of a sole -21 is limited. In a series
of 8 patients with a -21 as a sole abnormality, the median OS was
calculated as 32 months and the leukemia-free survival as 31.3 months,[2] which was in good accordance with the previous study of Haase et al..[1] Thus, an isolated -21 would fit into the intermediate risk group according to the IPSS-R.[7]
However, due to the very low number of patients, this prognostic
assessment needs to be considered with caution. Monosomy 21 is
associated with low peripheral blood values and elevated bone marrow
blasts. In 8 patients with an isolated -21, reported by Schanz et al.,
the median hemoglobin level was 7.2 g/dl, the median platelet count was
35/nl and the median ANC count 2.2/nl, respectively. The median bone
marrow blast count was 13%.[2] The finding of a -21 by
karyotyping, sometimes, does not reflect a real monosomy but a
technical artifact that mimics the respective abnormality. Loss of
chromosome 21 may occur randomly by preparing the cells for chromosome
banding analysis. Thus, the finding of a monosomy 21 by chromosome
banding analysis should be confirmed by other techniques like FISH at
least if it appears in a small mosaic.[32]
Chromosomal Gains
Trisomy 11. Isolated trisomy 11 (+11) is a very rare abnormality in MDS. The frequency was described between 0.2 and 0.3% of all MDS cases.[2,33-35] Wang et al.[33]
report on a retrospective study with a duration of 15 years including
17 out of 5,000 MDS patients with +11 as a sole abnormality (n=10) or
as part of a non-complex karyotype (n=7). The additional abnormalities
contained trisomies of chromosomes 2, 8, 10, 19 or 22 without any
typical poor-risk abnormalities such as chromosome 7 abnormalities. MDS
patients with trisomy 11 showed a median survival time of 14 months
only and a high frequency of disease progression. Within a median
interval of 5 months 69% of the affected patients developed s-AML, the
remaining patients progressed to advanced MDS stages.[33]
Trisomy
11 can also occur in patients with AML. MDS and AML patients with +11
were reported to show early stem cell features with a low degree of
maturation.[33,35,36] AML patients
with trisomy 11 as isolated abnormality or within a non-complex
aberrant karyotype were reported to show as well adverse outcomes with
a median OS of only 9 months.[33] Alseraye et al.[35]
published a retrospective study including more than 20,000 AML patients
in a 10-years-period and found 18 patients (0.09%) with isolated +11:
14 patients with de novo AML and 4 patients with s-AML following MDS
who showed a median OS of 5 months and an aggressive course of the
disease. Obviously, +11 does not show any specific association to a
certain WHO (WHO) or FAB MDS subtype or secondary or therapy-related
MDS.[33]
Trisomy 11 affects the MLL gene on 11q23.[37] Sixty-four percent of AML and 50% of MDS patients with +11 showed MLL partial tandem duplications (MLL-PTDs).[33] Twenty to twenty-five percent of AML with +11, but no MDS patients with this cytogenetic abnormality presented an additional FLT3-ITD mutation.[33,38]
Because
of the very low frequency of isolated +11 in MDS, it was so far
assigned to the intermediate risk category according to the IPSS and
the IPSS-R, respectively.[2,7,39]
According to the published literature, trisomy 11 seems to be
associated with an aggressive course of the disease, short OS, and
rapid progression or leukemic transformation.
Trisomy 13.
Trisomy 13 (+13), especially as solitary abnormality, is very rare,
however it has been observed recurrently in myeloid neoplasia, mostly
in AML, primary myelofibrosis, atypical CML and MDS.[40] Isolated +13 in a patient with MDS RAEB was first reported in 1989.[41] A second case report – also MDS RAEB - followed in 1990.[42]
In the same year +13 was identified as a rare but recurrent abnormality
in de novo acute leukemia being observed in 8/621 (1.3%) consecutive
patients (5 AML, 1 acute mixed lineage leukemia, 1 ALL, 1 acute
undifferentiated leukemia). The survival of these patients was poor
with a median of 9.5 months (range: 0.5 to 14.7 months).[43]
The United Kingdom Cancer Cytogenetics Group published a survey report
on patients with +13 and myeloid malignancy. Of the 28 patients
reported 5 (18%) had MDS (1 RA, 1 RAEB, 5 RAEB-t) while the others had
AML. In general the OS of the entire group was poor (median: 3.0
months, range: 1-96), however no prognostic data were available for
single patients with MDS.[44] In a multicenter
cytogenetic study our group observed isolated +13 in 4 (0.2%), with one
additional abnormality in 4 (0.2%) and in 8 (0.4%) patients as a
component of complex changes in a cohort of 2,072 patients with MDS.[1]
This is contrasting the statement in the “Huret Atlas” saying that in
the majority of cases +13 is the only cytogenetic abnormality.[40] In AML, trisomy 13 was shown to be closely associated with FLT3 overexpression and cooperating RUNX1 mutations.[45,46]
In
a large series published by the Mayo Clinic in Rochester +13 was
detected in 0.2% (n=9) of all MDS and 0.7% (n=15) of all AML patients.
Furthermore one patient with primary myelofibrosis, one with CMML and
one with pernicious anemia had +13, too. There was a clear gender
imbalance with 21/27 males. The median age was 73 years. In 96% of
these cases, +13 was isolated. In the 9 MDS cases, the majority showed
blast increase and disgranulopoiesis with hypogranulation. Furthermore,
6/9 patients showed small monolobated megakaryocytes remembering the
situation in del(5q) MDS. Two patients with higher risk MDS were
treated with hypomethylating agents but did not respond. Blood values
(medians) for the whole +13 cohort were Hb = 9.0 g/dl (6.2-13),
leukocytes = 5.3/nl (1.0-264.8) and platelets = 87/nl (11-312). The
whole trisomy 13 cohort (summarizing the MDS and AML patients) revealed
a median survival of 5.8 months (range: 0.5 – 24).[47]
In 2 older male patients with AML and isolated +13 treatment with high
dose lenalidomide resulted in a sustained morphologic and cytogenetic
complete remission in this otherwise poor risk cytogenetic subset of
AML not responding to standard high-dose chemotherapy.[48]
In
conclusion, +13 is a very rare condition in MDS, occurring in 0.2 to
0.8% of patients, mostly as an isolated change. In AML +13 is a bit
more frequent with an incidence between 1 and 2%. The majority of
patients have isolated +13. Most patients are over 70 years old and are
males. Typically, the MDS is advanced with blast excess and moderate
pancytopenia. Although most survival data relate to AML cases, the
prognosis is bad with median survival ranging between less than six
months and one year. Patients with AML and +13 do not respond to
standard intensive chemotherapy, and hypomethylating agents for MDS
patients also might be inefficient although database is not satisfying
concerning this issue. However, high-dose lenalidomide could be an
option. On a molecular level, +13 is associated with an overexpression
of FLT3 located at the triplicated chromosome. Furthermore, there is a
close association between +13 and mutations of RUNX1.
Trisomy 14/14q. Trisomy 14, in general, is extremely rare in hematologic malignancies but shows a clear association with myeloid neoplasia.[49]
Trisomy 14/14q has been mainly observed in MDS and CMML; more rarely it
has been seen in AML and in atypical MDS/MPN overlap syndromes.[49-52]
Most reports in the literature refer to single cases or smaller series
of patients. In the largest series (n=16) published so far, 10 patients
presented with isolated complete trisomy 14, in one case trisomy 14
represented the second evolutionary step in a patient with loss of the
Y-chromosome as primary change. Finally, 5/16 patients showed an
isolated isochromosome 14q – i(14)(q10) – for the long arm, resulting
in trisomy 14q. In this series, 7/16 patients had MDS (including 4
RAEB-1/-2, 4 CMML, 4 AML and one atypical MDS/MPN overlap syndrome).[50]
The median age was ranging between 67 and 72 years. There was a male
preponderance with roughly 70% of patients being of the male gender.
In
a multicenter cytogenetic study we observed +14 in 3 (0.1%) patients,
with one additional abnormality in 4 (0.2%) and in 9 (0.4%) patients
with complex changes out of 2,072 patients of the entire cohort.[1] To the best of our knowledge, this is the only published report[1]
on the frequency of trisomy 14 as yet. It has to be mentioned that
cases with i(14)(q10) were not included in our dataset. The cumulative
incidence combining trisomy 14 and i(14)(q10) may be one-third higher.
Hematologic
findings in patients with trisomy 14/14q are inconsistent. Mancini
reported a tendency to normal or increased platelets while in the case
collection of Cui patients with MDS showed a broad range of platelet
levels (23 – 465/µl, median 79/µl). Remarkably thrombocytosis was only
observed in 2 RARS cases, whereas in the remaining five MDS cases (4
RAEB-1/-2 and 1 RCMD) platelets were low to normal (23 – 156/µl, median
75/µl). Patients with CMML showed normal to increased platelet counts
(160 – 626/µl, median 198/µl). Hb values ranged between 4.0 g/dl and
12.0 g/dl (median 9.8 g/dl) in the review of Mancini with comparable
data (median 10.0 g/dl) in the series of Cui.[50,51]
Leukocytes ranged between 2.6/nl and 9.2/nl (median 3.8/nl) in
Mancini´s series and between 1.6 and 8.8/nl (median 4.2/nl) in Cui´s
cohort thus being moderately reduced to normal.
In a literature review, survival data from 18 patients suggest an intermediate prognosis.[51]
These data are supported by the findings of Cui et al. who reported a
median survival of 28 months in MDS, 31.5 months in MDS/MPN and nine
months in AML.[50]
The only systematic molecular investigation (KIT, RAS, FLT3, NPM1, JAK2) in this cytogenetic subgroup identified FLT3-ITD in 1/6 MDS at diagnosis later transforming to s-AML and KRAS-mutation
in 1/7 MDS patients at the time of leukemic transformation. Thus, the
molecular background in these cases remains to be clarified.[50] More comprehensive analyzes are needed, however surely hampered by the scarcity of trisomy14/14q.
Taken
together trisomy 14/14q occurs in less than 0.5% of patients with MDS
as isolated change. The abnormality seems to be an early event, and the
affected cell clones seem to be genetically stable with a low tendency
to acquire additional changes and a low tendency to leukemic
transformation.[50] The abnormalities typically can
be seen in older male MDS patients with and without blast excess. The
prognosis seems to be intermediate to good. Blood counts typically show
normal leukocytes or mild leukocytopenia, moderate anemia and
moderately reduced to normal platelets with the exception of
thrombocytosis in RARS. In CMML, platelets are not decreased. The
molecular background is unclear. Especially the role of oncogenes
located at 14q needs further elucidation.
Trisomy 21.
Trisomy 21 (+21) is well known in the context of Down’s syndrome,
associated with a marked risk to develop AML during childhood. However,
besides this hereditary disease, +21 may also occur as a clonal somatic
abnormality in several hematologic disorders. In adult de novo AML,
trisomy 21 occurs in around 3% of patients.[53] In MDS, +21 as a single abnormality (Figure 2)
is occurring more rarely. In a series of 968 patients published by Solé
et al., isolated +21 was detected in 0.8% of patients and showed a
significant association with CMML, where 3.5% of patients showed +21 as
sole abnormality.[54] Further publications found a
comparable incidence, calculated as 1.1% of patients showing +21 within
a non-complex karyotype. Based on a cohort of 2,901 patients, the
incidence of +21 as an isolated abnormality was 0.3%, assigning this
abnormality to the group of rare abnormalities in MDS. Based on nine
patients, the authors described an association with a low ANC (median
1.9/nl) and a slightly decreased platelet (median 105/nl) and
hemoglobin (9.1 g/dl) level. The median blast count in these patients
was 6%, indicating an association with higher risk MDS.[2] The median OS was 100.8 months in +21 within a non-complex karyotype.[1] Other publications stated a median OS of 13.9 months[54]
and 21.5 months 2 respectively, for patients showing isolated +21. The
median time to AML evolution was 100.7 months in the publication of
Schanz et al..[2] Solé et al. stated a cumulative AML risk of 25% after one year and 50% after five years.[54]
Taken these results together, the prognostic impact of an acquired,
isolated +21 in patients with MDS remains unclear und has to be stated
as intermediate until a higher number of patients were analyzed.
The molecular background of patients showing +21 in myeloid malignancies remains undefined as yet. RUNX1 (=AML1), located on chromosome 8q22, commonly involved in t(8;21)/RUNX1-RUNXT1 in AML, was shown to be also point mutated in patients with myeloid malignancies like AML, MDS and MPN with +21.[55] A Japanese group found a poor prognostic impact of intragenic RUNX1 mutations in MDS but did not describe a correlation with +21.[56]
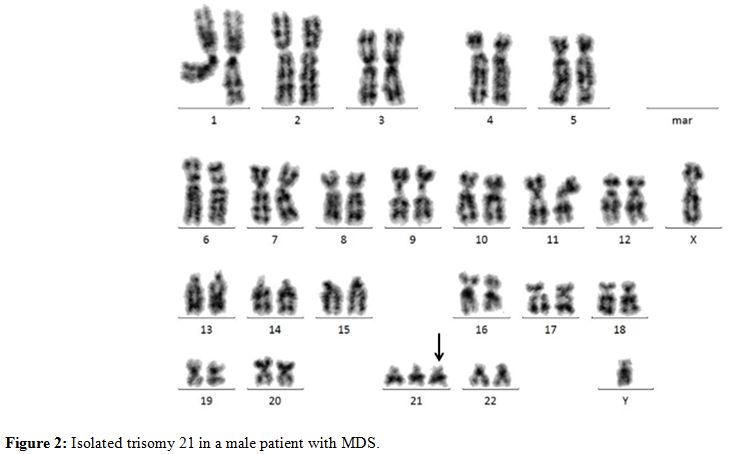 |
Figure 2. Isolated trisomy 21 in a male patient with MDS. |
Structural Abnormalities
Gain of chromosome 1q. Gain
of 1q resulting in complete or partial trisomy 1q can be due to
duplications, to the formation of an isochromosome i(1q), to the gain
of a deleted or a whole chromosome, or to unbalanced translocations
with different partner chromosomes as demonstrated in an MDS case
series presented by Fonatsch et al.[57]
Isochromosome 1q [i(1q)] may occur in different hematological malignancies such as ALL or lymphoproliferative disorders, or in solid tumors.[58] In myeloid disorders, i(1q) shows very rare occurrence.[58]
Pawarode et al. reported on a male patient developing t-MDS with
isolated i(1q) 14 years after therapy of acute promyelocytic leukemia
(APL). There was no blast increase, and the t-MDS showed only slow
progression under supportive therapy.[58] Occasional cases with i(1q) as isolated abnormality have also been reported as de novo MDS, e.g. a 20-year-old female patient with a hypocellular bone marrow who received allogeneic HSCT.[59]
Fonatsch et al. reported on a 24 years old male patient with RAEB-T and
isolated i(1q) who remained clinically stable over ten months.[57] Prognostic assessment of i(1q) is difficult due to the rare occurrence.
Duplication of 1q mainly
occurs as a secondary event in MDS. Occasionally, dup(1)(q21q32) has
been documented as a sole cytogenetic abnormality in MDS patients.
Alfaro et al. reported on two MDS patients with a sole dup(1)(q21q32).
One patient showed a clonal cytogenetic evolution with an additional
+8; the other patient evolved to s-AML.[60] The authors considered duplication of 21q to be prognostically adverse in MDS.
Furthermore, various unbalanced translocations
may lead to a gain of 1q. Derivative translocation der(1;5) was
described mostly in patients with AML, but also in patients with CML,
MPNs, or MDS. The breakpoints of der(1;5) vary from 1q11 to 1q43, with
a clustering to 1q21-23, and the 5q breaks occurred in 5q11 to 5q35,
with preponderance in the distal 5q3 region. Derivative translocation
der(1;5) was either reported as isolated abnormality or in combination
with other cytogenetic abnormalities.[61] According
to a collection of two own cases and a review of the literature,
Johansson et al. suggested that the prognostic impact of the respective
abnormality was poor as most patients included in this report had died.[61]
Lunghi et al. summarized several MDS cases with der(1)(t;16) from the
literature. Male preponderance was evident. Patients showed different
MDS subtypes with or without blast increase, but a high transformation
rate to s-AML was noted in 4 out of 7 patients. As shown by FISH
analyzes, the respective translocation was resulting in a gain of 1q
and loss of 16q.[62] Another rare unbalanced translocation in MDS resulting in a gain of 1q is der(1)(1;13)(q21;p12).[57]
Hypothetically,
the major consequence of these unbalanced translocations is the
chromosomal imbalance resulting from the gain of the long arm of
chromosome 1q and the loss of genetic material from the partner
chromosome rather than the chromosomal break per se.[62]
This argument is further underlined by the fact that the breakpoints
within 1q show considerable variation depending on the translocation
partners. Based on the analysis of a large cohort of 968 patients with
MDS, Sole et al. came to the conclusion that MDS with 1q involvement
shows a poor outcome when compared with the overall series of patients.
However, due to the rare occurrence of this abnormality the authors
recommended caution with regards to this interpretation.[54]
The most frequently reported unbalanced translocation in this context is the derivative translocation der(7)t(1;7)(q10;p10)
that represents an unbalanced whole-arm translocation between the
centromere-near regions of 7p and 1q and results in unbalanced gain of
1q and loss of 7q (Figure 3).
It mostly occurs in MDS, but may also occur in AML (often of secondary
origin) or myeloproliferative neoplasms (MPNs) such as myelofibrosis.
The der(7)t(1;7)(q10;p10) has been described to occur in 1-3% of MDS
cases, more frequently in males.[63,64] In half of
all cases, der(7)t(1;7)(q10;p10) occurs as sole abnormality, in a third
of cases it is accompanied by numerical gains such as +8 or +21;
additional structural abnormalities were described in around 15% of
affected cases.[63] Even in case of multiple
cytogenetic subclones, it could be shown that der(1;7) affects all
abnormal metaphases. Furthermore, the der(1;7) can be identified in all
affected patients already at first diagnosis of the disease[65] which suggests a high level of genetic stability for the respective cytogenetic abnormality.
A
poor prognostic impact and a high risk of transformation to s-AML has
been described in MDS patients carrying der(1;7). Slovak et al.
compared the prognosis of der(1;7) positive MDS patients with patients
with chromosome 7 abnormalities (-7 and 7q-; both are known to be
prognostically highly adverse) and found no significant differences
with regard to leukemic transformation rate or 5-year survival between
these different cytogenetic risk groups.[64] On the
other hand, Sanada et al. retrospectively compared a cohort of 77
patients suffering from different myeloid malignancies (MDS, AML, MPNs)
all carrying der(1;7) with patients with -7 or 7q. The MDS patients
with a der(1;7) showed significantly lower blast counts, higher
hemoglobin levels, and slower progression to s-AML as compared with the
cases carrying other chromosome 7 abnormalities.[65] However, as the median survival of the MDS patients with a der(1;7) was only 23.7 months in this study[65]
there was no doubt that the respective abnormality confers an adverse
prognosis in patients with MDS. Our unpublished data confirm the
significant better outcome of MDS patients with a der(1;7) as compared
with patients with -7 or 7q-.[66] Hsiao et al.
documented frequent occurrence of previous chemo- or radiotherapy or
additional cytogenetic changes in patients with MDS or AML with
der(1;7).[67] Westman et al. described an increased frequency of isocitrate dehydrogenase (IDH1 and IDH2) mutations in t-MDS/AML patients with der(1;7) and other +1q abnormalities.[68]
11q23/MLL translocations. Reciprocal translocations of 11q23, involving the MLL gene, are frequent in adult and pediatric acute lymphoblastic and myeloid leukemia. Rearrangements of the MLL
gene with several partner genes result in various effects on
hematopoietic stem cells, especially differentiation anomalies.
Actually, more than 70 translocation partners have been identified.[69] On the molecular level, the MLL gene may also be affected by partial tandem duplications (MLL-PTDs). 11q23/MLL
translocations are seen in around 22% of patients with ALL which is
much more frequent as compared to AML with only 5% of patients being
affected.[70] In patients with ALL, t(4;11) is most
frequent, followed by t(11;19). In AML, t(9;11) and t(10;11) are most
commonly detected.[70] In therapy-related AML/MDS, 11q23/MLL translocations are strongly associated with a previous exposition to topoisomerase-II inhibitors (e.g. etoposide).[71] About 5% of all patients showing 11q23/MLL translocations suffer from myelodysplastic syndromes.[72] 11q23/MLL translocations are very rare in primary MDS and can be found in 0.5% of patients as part of a non-complex karyotype.[1] The frequency of 11q23/MLL translocations as sole aberrations in MDS is even lower with 0.2% of all patients.2 11q23/MLL translocations are correlated with the WHO subtypes RA or RAEB-1.[72] In the 7 patients with sole 11q23/MLL
translocations described by Schanz et al., these abnormalities were
associated with a low hemoglobin level (7.9 g/dl) but a normal platelet
(140/nl) and ANC (6.3/nl) count. The median bone marrow blast count in
these seven patients was 4.0%. A higher frequency of 11q23/MLL translocations was described by Solé et al. in a study based on 968 patients with MDS. In this cohort, sole 11q23/MLL translocations were described in 6% of patients.[54] The authors found no correlation with FAB subtypes.
Regarding the prognostic impact of 11q23/MLL
translocations in MDS, Solé et al. found a median OS of 26 months and a
cumulative risk for developing acute leukemia of 40% to 1 year and 92%
to 5 years,[54] but did not discriminate between the different types of 11q23/MLL translocations. In another study, the median OS of patients with 11q23/MLL translocations occurring within a non-complex karyotypes was 20 months.[1]
In this study, primary as well as therapy-associated MDS patients were
included. The largest series of patients with a primary, untreated MDS
described a median OS of 26.7 months and a median AML-free survival of
78 months.[2] However, also due to the low number of patients showing 11q23/MLL
translocations in MDS, their real prognostic impact is not well defined
for this hematologic entity. Consequently, as other rare single
abnormalities, 11q23/MLL translocations were assigned to the intermediate risk group in the IPSS and also in the IPSS-R.[7,39]
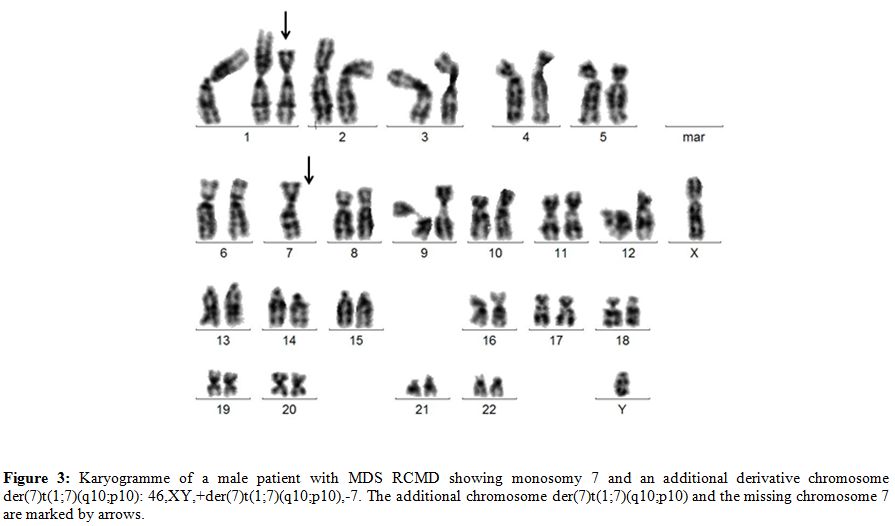 |
Figure 3. Karyogramme of a male patient with MDS RCMD showing monosomy 7 and an additional derivative chromosome der(7)t(1;7)(q10;p10): 46,XY,+der(7)t(1;7)(q10;p10),-7. The additional chromosome der(7)t(1;7)(q10;p10) and the missing chromosome 7 are marked by arrows. |
Conclusion
Due to the missing statistical validity caused by the low number of
cases, rare cytogenetic abnormalities that are not considered
specifically by the IPSS-R are assigned to the prognostically
intermediate cytogenetic subgroup[2] corresponding to 2.0 scoring points within this risk stratification system.[7] Although the cytogenetic scoring categories of the IPSS-R include already more than 90% of patients with MDS,[2]
several patients remain for whom a correct cytogenetic classification
is not available so far. Although a complete overview of all relevant
rare cytogenetic abnormalities in MDS1 is beyond the scope of this
review, this article provides examples for cytogenetic abnormalities
that could be integrated into risk stratification systems. According to
the above-cited studies and own experience, loss of the X-chromosome,[2] loss[1,2] and gain[2,54]
of chromosome 21, and 11q23/MLL translocations are associated with an
intermediate prognosis. Trisomy 14 is prognostically intermediate to
favorable.[50,51] 13q deletion is associated with favorable response to immunosuppressive therapy.[29,30] The prognostic impact of a der(7)t(1;7)(q10;or p10) is less adverse as compared to monosomy 7 or 7q deletion.[66] Trisomy of chromosomes 11[33] and 13[47]
mediates an unfavorable impact. These and other examples of
prognostically relevant rare cytogenetic abnormalities emphasize the
necessity to expand existing prognostic models and to optimize the
IPSS-R.[2,7]
Finally, our
working group would like to emphasize our interest in collecting
additional cases with rare cytogenetic abnormalities with the aim to
expand our database, to increase insights in the cytogenetic complexity
of myelodysplastic syndromes, and to promote the exchange between
hematologists interested in this issue.
Acknowledgements
The authors would like to thank Dr. Christina Ganster and Dr. Katayoon Shirneshan from the Department of Hematology and Medical Oncology, Medical Center Göttingen, for providing the figures for this article..
References
 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . [TOP]