Received: April 26, 2015
Accepted: June 13, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015043, DOI 10.4084/MJHID.2015.043
This article is available on PDF format at:

Uday Kulkarni1, Anna Valson2, Anila Korula3 and Vikram Mathews1
1 Department of Clinical Haematology. Christian Medical College and Hospital, Vellore, Tamil Nadu, India.
2 Department of Nephrology. Christian Medical College and Hospital, Vellore, Tamil Nadu, India.
3 Department of General Pathology. Christian Medical College and Hospital, Vellore, Tamil Nadu, India.
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract We describe the first case from India
of ALECT2 amyloidosis. An adult Punjabi male presented with progressive
renal dysfunction and non-nephrotic range proteinuria. Serum protein
electrophoresis and immunofixation were normal, with mildly elevated
serum free light chain ratio. A renal biopsy confirmed the presence of
amyloid. Immunohistochemistry was negative for monoclonal light chains.
Proteomic analysis confirmed the presence of ALECT2 amyloid. The
present case highlights the need for confirmatory testing for typing of
amyloid. |
Case
A 52 year old Punjabi male was evaluated elsewhere for bilateral leg
swelling and reduced urine output. He was noted to have renal
dysfunction and underwent a renal biopsy that was consistent with
amyloidosis. Echocardiography revealed left ventricular diastolic
dysfunction. Bone marrow revealed 10% plasma cells. Serum protein
electrophoresis and immunofixation were normal. Free light chain ratio
was mildly elevated (1.86). Whole body PET-CT (positron emission
tomography-computed tomography) did not reveal any lytic lesions. He
was diagnosed to have hypertension three years ago and was well
controlled on oral cilnidipine. He was referred to our hospital as AL
amyloidosis for autologous bone marrow transplantation.
At our
hospital, general and systemic examination was unremarkable. His blood
pressure was 130/80 mmHg. The renal biopsy was reviewed. It showed
Congo red positive pale, amorphous, acellular, eosinophilic mesangial
deposits consistent with amyloid. However, the immunohistochemistry was
not conclusive. His creatinine was 1.47mg% and 24 hour urine protein
was 99 mg. Urine Bence-Jones protein was negative. Serum protein
electrophoresis and immunofixation were normal. Serum free light chain
ratio was 4 (kappa 40mg/L; lambda 10mg/L). Review of the PET-CT did not
reveal any lytic lesions. There was no anemia or hypercalcemia. Bone
marrow was mildly hypercellular with nonspecific reactive changes.
Echocardiography was normal.
He underwent a repeat renal biopsy (Figure 1)
which showed glomerular, megangial and capillary wall deposits of pale,
acellular, eosinophilic material staining for amyloid with Congo Red
and Thioflavine T histochemical stains. The interstitium and
extraglomerular blood vessels showed similar deposits.
Immunohistochemistry for monoclonal kappa and lambda light chains was
negative. As the patient was referred to our hospital with a diagnosis
of AL amyloidosis, the paraffin block was sent for proteomic analysis
for exact typing of the amyloid. Liquid chromatography tandem mass
spectrometry was performed at Mayo Clinic Laboratories, USA. The
peptides were extracted from Congo Red positive microdissected paraffin
embedded renal tissue. Mass spectrometry detected a peptide profile
consistent with leucocyte-derived chemotaxin-2 type amyloidosis. Since
there is no established treatment modality for ALECT2 amyloid, regular
monitoring of renal function and blood pressure control were advised.
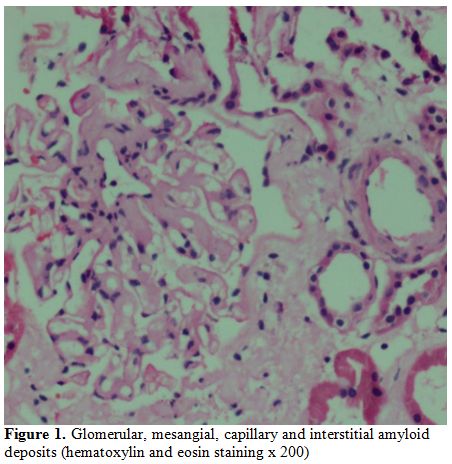 |
Figure 1. Glomerular, mesangial, capillary and interstitial amyloid deposits (hematoxylin and eosin staining x 200). |
Discussion
ALECT2, first reported in the year 2008, is one of the most recently described types of amyloidosis.[1] It is the third most common type of systemic amyloidosis after AL and AA.[2]
LECT2
is a normal serum protein synthesized by the liver that is chemotactic
for neutrophils, and a growth factor for chondrocytes and osteoblasts.
However, the plasma levels of this protein are not increased in ALECT2
amyloidosis, and there are no identified mutations in LECT2 causing misfolding leading to amyloidosis. A common G/G polymorphism at position 172 in LECT2
leading to the replacement of isoleucine by valine may bring about a
conformational change that predisposes to amyloidogenesis. However, a
current theory suggests that it is a digenic disease, requiring a
second mutation at an as yet unidentified locus.[3]
ALECT2
amyloidosis primarily involves the kidney, but liver, spleen, lung and
adrenal involvement have also been described. It is common in certain
ethnic groups like Hispanics, Arabs, and Punjabis.[4,5]
The usual presentation is of an elderly individual presenting with
progressive renal insufficiency with or without proteinuria, which is
usually non-nephrotic, unlike AL and AA amyloidosis, in which
proteinuria is typically in the nephrotic range.[5,6]
Many of these patients also have a concomitant monoclonal gammopathy of
unknown significance. Hence, it is important to distinguish these cases
from AL amyloidosis to avoid unnecessary and often harmful
chemotherapy.[3]
Although immunohistochemistry is
a useful tool for confirming the type of amyloid, more accurate
techniques like mass spectrometry based proteomic analysis are required
in inconclusive cases.[7] While anti-LECT2 antibody on
immunohistochemistry can localize this form of amyloidosis to
glomerular, interstitial and vascular deposits; liquid chromatography
tandem mass spectrometry is used to confirm the diagnosis and exclude
uncommon and familial forms of amyloid.
A concurrent renal
disease like diabetic nephropathy and IgA nephropathy is common.
Because cardiac involvement is rare, patient survival is superior to AL
and AA amyloidosis. Median renal survival is 62 months in those without
a concurrent renal disease. There is no available treatment for ALECT2
amyloidosis, except renal transplantation once end stage kidney disease
is established.[8]
The present case highlights the following points:
1.
Gradually progressing renal dysfunction even with a non-nephrotic range
proteinuria may be a manifestation of renal amyloidosis.
2. When
amyloid is noted histopathologically, the presence of a monoclonal
gammopathy does not confirm the type of amyloid as AL.
3.
The presence of predominantly renal interstitial and mesangial deposits
of amyloid should alert one to the possibility of ALECT2 amyloidosis.
4.
If immunohistochemistry is negative, mass spectrometry based proteomic
analysis is necessary for confirming the type of amyloid.
5. ALECT2 amyloid may be an important cause of amyloid in India, especially among Punjabis.
6.
It is important to establish this diagnosis to avoid chemotherapy or an
autologous stem cell transplantation that may be considered in the
absence of defining the type of amyloid.
Acknowledgments
The authors wish to thank Mayo Clinic Laboratories, USA for the results of the proteomic analysis.
References
 http://dx.doi.org/10.1038/ki.2014.11
http://dx.doi.org/10.1038/ki.2014.11 [TOP]