Received: January 1, 2015
Accepted: June 15 , 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015046, DOI 10.4084/MJHID.2015.046
This article is available on PDF format at:

Sudhansu Sekhar Nishank
Division of Genetics, Regional Medical Research Centre for Tribals (ICMR), Nagpur Road, PO-Garha, Jabalpur-482003, Madhya Pradesh, INDIA.
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Background: Oxidative stress
constitutes one of the significant causes of vaso-occlusive clinical episodes
in sickle cell disease (SCD) patients. It brings about the generation of
reactive oxygen species and consequent damage to DNA. DNA damage repair genes
such as hOGG1, XRCC1 and p53 play an important role in the repair
of DNA damage during oxidative stress. However, it is not known as to the role
of these genes in oxidative stress mediated vaso-occlusive clinical
complications of SCD patients. Objective: To see the possible
association of DNA repair gene polymorphisms with clinical manifestation of SCD
patients. Methods: Genotyping of DNA damage repair
genes by PCR-RFLP, measurement of oxidant and anti-oxidant status, along with a
clinical evaluation of 250 SCD patients and their comparison with normal
individuals. Result: The level of oxidants was
high, and that of antioxidants was low in SCD patients compared to normal
individuals. The prevalence of mutant alleles of hOGG1 gene, XRCC1 gene
(codon 280 Arg>His) were found to be significantly higher among SCD patients
as compared to controls. However, SCD patients did not show clinical
association with any of these DNA repair gene polymorphisms. Conclusion: This indicates that hOGG1,
p53and XRCC1 gene polymorphisms have no clinical association with
SCD patients in India. |
Introduction
Sickle cell disease (SCD) patients (homozygous sickle cell, Hb SS) are usually associated with activation of enzymatic and non-enzymatic sources of reactive oxygen species (ROS) leading to oxidative stress. The resultant oxidative stress leads to microvascular dysfunction, tissue injury and vaso-occlusive crisis led morbidity and mortality among SCD patients.[1] In higher eukaryotes, DNA is prone to oxidative damage by ROS, which leads to replication errors and genome instability. This oxidative base damage is highly mutagenic, and if unrepaired it produces life threatening malignancies in human. Sickle cell traits and sickle cell homozygotes in many parts of the world have also been found to be associated with malignancies such as renal medullary carcinoma, multiple myeloma, osteosarcoma, malignant fibrous histiocytoma in the distal tibia.[2,3,4] The DNA damage repair genes, such as hOGG1(human 8-oxoguanine DNA glycosylase 1) gene, X-ray repair cross complementing group1(XRCC1) and tumor suppressor gene p53, play important role in repair of DNA damage caused by oxidative stress during DNA replication. Polymorphisms of hOGG1 Ser>Cys, p53 Arg>Pro and XRCC1 (codon 194 Arg>Trp, codon 280 Arg>His, codon 399 Arg>Gln) have been variably associated with repair of oxidative stress mediated DNA damage in normal human.[5] Transfused sickle-β-thalassemia patients who did not undergo chelation shows significant damage to DNA due to oxidative stress compared to normal individuals.[6] Sickle cell disease patients treated with hydroxyurea showed a higher level of DNA damage than controls irrespective of gender, smoking or age.[7] Oxidative stress promotes vaso-occlusive events and consequent clinical complications in SCD patients. It is known that DNA damage repair genes (enzymes) act as sensitive biomarkers of DNA damage repair efficiency of the cell.[8] Although oxidative stress that promotes damage to DNA brings about endothelial dysfunction leading to vaso-occlusive complications in SCD patients, it is likely that there is a relationship between DNA repair gene polymorphism and clinical complications in SCD patients. Thus, the present study aims to find out the possible association of DNA repair gene polymorphisms and clinical symptoms of SCD patients belonging particularly to the state of Madhya Pradesh (India).
Materials and methods
The study sample involved about 250 SCD patients (homozygous sickle
cell, Hb SS) and250 normal individuals (having phenotype Hb AA) from
unrelated families belonging to the state of Madhya Pradesh (India).
The samples were collected at the sickle cell clinic of Regional
Medical Research Centre for Tribals (ICMR), in the campus of NSB
Medical College Jabalpur, Madhya Pradesh (India). The study sample
included individuals belonging to Scheduled Caste (SC), Scheduled Tribe
(ST) and general population of state of Madhya Pradesh, where both SCD
patients and normal individuals were matched with each other with
respect the age and ethnicity. Among the study samples, 120 females,
130 males were SCD individuals whereas 118 females and 132 males were
normal individuals.
The study was initiated after obtaining
written consent of patients/their parents and approval by an ethical
committee of RMRCT (ICMR), India and NSB Medical College. The patients
having recent or multiple blood transfusions and infection and pregnant
women were excluded from the study. Besides the present study was
conducted in accordance with ethical standards of Helsinki Declaration.
Clinical complications such as chest pain, abdominal pain, bone joint
pain, history of blood transfusion, the degree of splenomegaly,
frequency of blood transfusion were recorded from the date of onset of
disease.
Oxidant and anti-oxidant status of sickle cell disease
and normal individuals were assessed by measurement of plasma/serum
level of Vitamin C, Vitamin E, 8-OHdG (8-hydroxy-2’-deoxyguanosine),
reduced glutathione (GSH), malondialdehyde (MDA), albumin, activity of
lactate dehydrogenase (LDH) and albumin using commercially available
Elisa kits (Sigma Aldrich, USA; Cell Biolab, USA) and automated
biochemical analyzer wherever necessary. Hemoglobin level was measured
by an automated blood cell counter (Sysmex Corporation, Japan).
Genotyping
of all DNA repair genes gene was performed by PCR - Restriction
Fragment Length Polymorphism technique as per published
procedure.[9-12] The p53 codon Arg72Pro was genotyped by using primers
– 5’ATC TAC AGT CCC CCT TGC CG3’ and 5’ GCA ACT GAC CGT GCA AGT CA3’. A
296 bp PCR product (denaturation at 950C 5 min, 35 cycles of 40 s at
95°C, 40 s at 60°C and 40 s at 72°C) was cleaved by Bst UI enzyme
(New England Bio Lab) and run in 2% agarose gel.[9] Homozygous Arg/Arg
individuals had two fragments of 169 and 127 bp. Homozygous Pro/Pro
individuals had a single fragment of 296 bp, and heterozygous Arg/Pro
individuals revealed all three fragments. In case of XRCC1 gene, Arg to
Gln substitution in exon 10 (codon 399) was amplified to form an
undigested fragment of 242 bp using primer pairs 5’ CCC CAA GTA CAG CCA
GGT 3’ and 5’ TGT CCC GCT CCT CTC AGT AG3’. The PCR product was
digested with Msp1 and
analyzed in 2% agarose gel. Homozygous Gln-Gln individuals reflected a
single product of 242 bp, homozygous Arg-Arg individuals demonstrated
both 148- and 94-bp fragment whereas Arg-Gln individuals revealed all
three of the fragments.[9] For Arg to Trp substitution in exon 6 (codon 194 of XRCC1
gene), a 485 bp PCR fragment was obtained using primer pairs 5’ GCC AGG
GCC CCT CCT TCA A3’ and 5’TAC CCT CAG ACC CAC GAG T3’. The PCR products
were digested with PVU II and
analyzed in a 2% agarose gel. Homozygous Arg-Arg individuals reflected
a single product fragment of 485 bp whereas homozygous Trp-Trp
individuals demonstrated both 396- and 89- bp fragments and
heterozygous Arg-Trp individuals revealed all three of fragments.[10] For XRCC1 Arg280His codon, a PCR product of 304 bp fragment demonstrates homozygous dominant whereas digestion of PCR product by Rsa I
into 246 bp- and 58 bp fragments demonstrate homozygous mutant. Primers
used for it were 5’CCC CAG TGG TGC TAA CCT AA3’ and 5’CTA CAT GAG GTG
CGT GCT GT3’.[11] For XRCC1 gene, PCR conditions were denaturation at 95°C 5 min, 35 cycles of 40 s at 95°C, 40 s at 60°C and 40 s at 72°C). Genotyping of hOGG1
Ser 326 Cys was done by PCR-RFLP involving primers – 5’TTG CCT TCG GCC
CTG TTC CCC AAG GA3’,5’ TTG CTG GTG GCT CCT GAG CAT GGC CG3’ and
restriction enzyme Msp I. The PCR product was 168 bp and Msp I digested products were 142, 26 bp for homozygous mutant. The PCR condition was same as described for XRCC1 gene except the annealing temperature kept at 65.5°C.[12]
Statistical analysis for Fisher’s exact X2
test using the odds ratio (OR), 95% confidence interval was performed by
statistical software Graph Pad Prism version 5.0 (La Jolla, CA, USA).
The X2 test was also used to test the Hardy-Weinberg equilibrium among the study subjects. The value of P < 0.05 was considered to be significant.
Results
The mean age of SCD patients was 16.3 (±6.0,Standard Deviation) years, and that of the control group was 17.6 (±6.8, SD) years. The frequency of mutant alleles hOGG1 326Cys (51.8%) and XRCC1 280 His (54.8%) were found to be significantly high in SCD individuals as compared to normal individuals (33.8% and 36.4% respectively). On the other hand there were no significant differences in the frequency of XRCC1 codon 194 and codon 399 alleles as well as mutant allele of p53 genes (Table 1 and Table 2) between SCD and normal individuals. Comparison of oxidant and antioxidant status of SCD and normal individuals showed that SCD patients had significantly lower level of antioxidants such as Vitamin C, Vitamin E, albumin as compared to controls. On the other hand, SCD individuals had significantly higher level of oxidants such as 8-OHdG and MDA along with a lower level of GSH along with lower activities of LDH (Table 3). There were no difference among the SCD patients with and without mutations in hOGG1 326Cys and XRCC1 280His alleles with respect to yearly incidence of chest pain, bone joint pain, fatigue, fever, blood transfusion along with degree of splenomegaly and age onset of disease (Table 4).
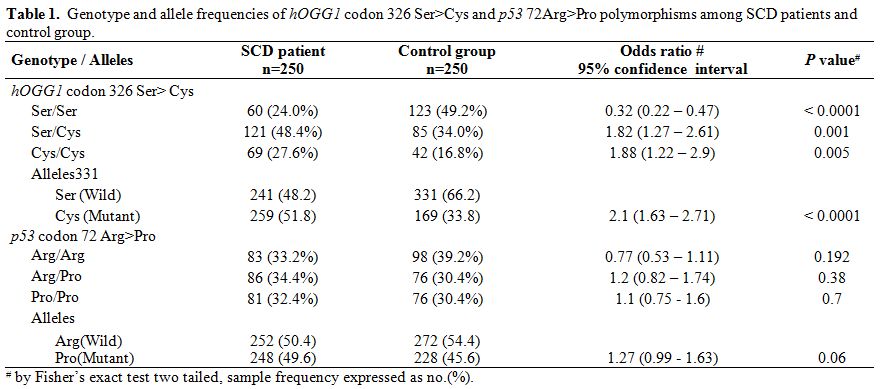 |
Table 1. Genotype and allele frequencies of hOGG1 codon 326 Ser>Cys and p53 72Arg>Pro polymorphisms among SCD patients and control group. |
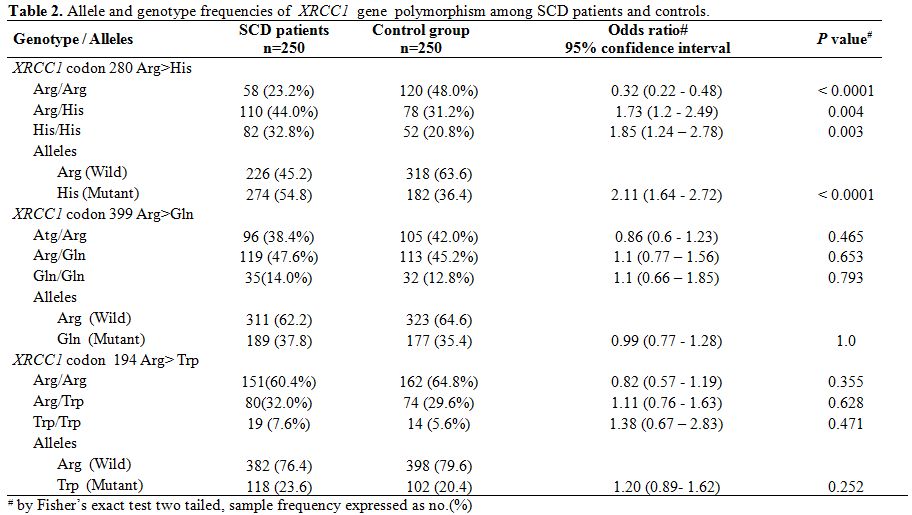 |
Table 2. Allele and genotype frequencies of XRCC1 gene polymorphism among SCD patients and controls. |
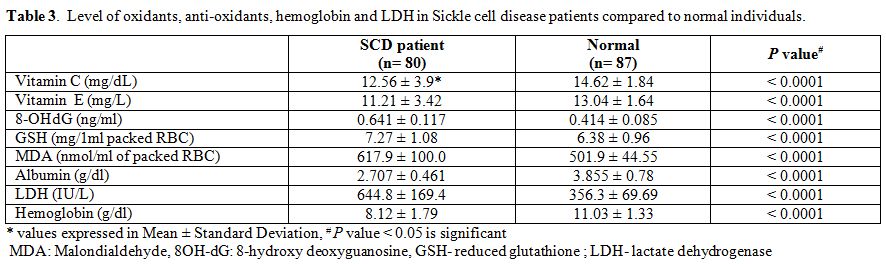 |
Table 3. Level of oxidants, anti-oxidants, hemoglobin and LDH in Sickle cell disease patients compared to normal individuals. |
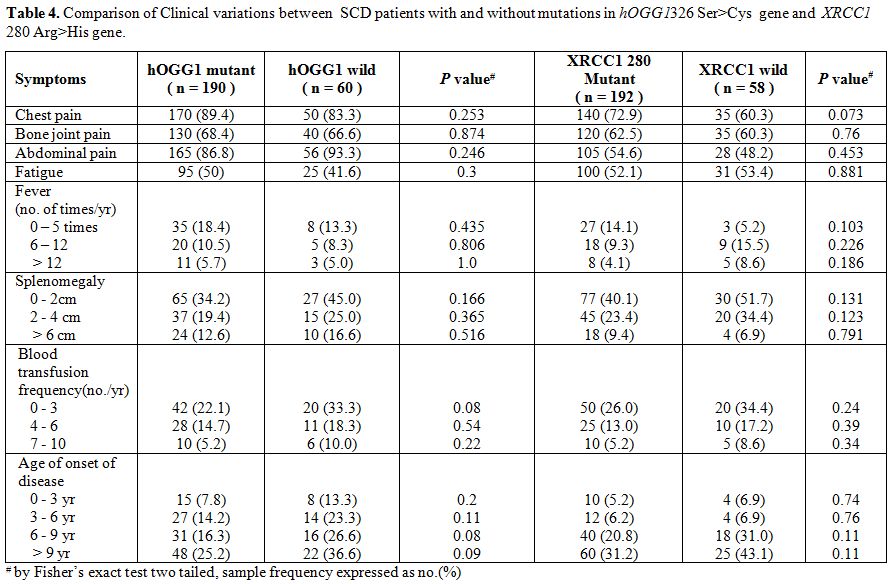 |
Table 4. Comparison of Clinical variations between SCD patients with and without mutations in hOGG1326 Ser>Cys gene and XRCC1 280 Arg>His gene. |
Discussion
Clinical complications of sickle cell disease (SCD) involve
generation and impairment of oxidative stress. There is a relationship
between markers of oxidative stress and common secondary diseases in
SCD such as acute chest syndrome and pulmonary hypertension.
Autoxidation of sickle hemoglobin (HbS) along with repeated cycle of
sickling and unsickling cause premature destruction of erythrocytes and
generation of reactive oxygen species (ROS) leading to oxidative
stress.[1] This is evidenced in the present study
which shows low level of antioxidants (such as Vit C, Vit E, albumin,
GSH) and high level of oxidants (such as 8OHdG, MDA) in SCD patients
compared to normal individuals. The lower level of antioxidants
particularly Vit C, Vit E, albumin, GSH along with higher level of LDH,
low level of hemoglobin in this study are supported by earlier findings
in SCD patients.[13] The high level of MDA in SCD
patients observed in the present study is similar to earlier
observation of higher level of MDA produced in related hemoglobinopathy
patients particularly beta thalassemia patients of India.[14]
Oxidative products such as MDA and 8-OHdG are known to be mutagenic and
cause damage to DNA. Besides low levels of antioxidants such as Vitamin
E and Vitamin C are found to cause genome instability and damage DNA.[15]
Both of these events may promote carcinogenesis if these oxidants
accumulate in the cells. However, DNA repair genes play a significant
role in the repair of DNA damage caused by oxidants. It is observed
that combination of different variants in DNA damage repair enzymes may
modulate the production of 8-oxoguanosine adducts in white blood cell
exposed to mutagens.[16] Similarly individuals carrying XRCC1 280 His allele and hOGG1
326Cys allele have an increased risk of chromosomal aberrations and
many individuals having these mutant alleles have higher incidence of
malignancy in normal population.[17,18,19] Although the frequency of these mutant alleles of XRCC1 and hOGG1
genes is found to be elevated in a present study of SCD patients; these
alleles seem incapable of explaining the usual clinical complications
of SCD patients. Rather, adhesion of the sickle cell to endothelium due
to oxidative stress cause inflammation that leads to the major cause of
clinical symptoms in SCD.[13]
Therefore, it
appears that there is absence of association between DNA repair gene
polymorphisms and clinical symptoms, as reflected in the present study.
Frequency of XRCC1 280 His
allele in normal individuals of the present study is different from the
normal population in Asian (7 to 15%), African (3%), Caucasians (5 to
9%). Similarly, frequency of hOGG1
326Cys allele is different from other countries (39 to 74% in China,
20.2% in Caucasians) including earlier Indian study (28 to 29%).[20,21] This discrepancy in findings may imply variation in ethnicity, environmental factors.
However,
further study of SCD populations from different communities of India
may give insights on the role of DNA damage repair gene polymorphisms
in clinical manifestation of SCD patients. Thus given the present study
findings, it may be concluded that hOGG1, XRCC1, and p53 gene polymorphisms do not seem to play a significant role in clinical manifestations of SCD patients of India.
Conclusion
There are no significant differences in the distribution and clinical impact of hOGG1, p53 and XRCC1 gene polymorphisms among SCD patients in India.
Acknowledgement
References
 .
.