Received: September 9, 2015
Accepted: October 10, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015060, DOI 10.4084/MJHID.2015.060
This article is available on PDF format at:

Said Y. Alkindi1, Anil Pathare2, Shoaib Al Zadjali2, Vinodhkumar Panjwani2, Fauzia Wasim2, Hammad Khan2, Pradeep Chopra2, Rajagopal Krishnamoorthy3 and Salam Alkindi4*
1 Ministry of Health, Muscat, Oman & McMaster University, Canada.
2 Sultan Qaboos University Hospital, Muscat, OMAN.
3 INSERM, U665, F-75015 Paris, France; Laboratoire d’Excellence GR-EX, Paris, France.
4 Sultan Qaboos University, College of Medicine & Health Sciences, Muscat, OMAN.
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Background: We explored the
potential relationship between steady state serum bilirubin levels and
the incidence of cholelithiasis in the context of UGT1A1 gene A(TA)nTAA
promoter polymorphism in Omani sickle cell anemia (SCA) patients,
homozygotes for African (Benin and Bantu) and Arab-Indian βS haplotypes, but sharing the same microgeographical environment and comparable life style factors. Methods: 136 SCA patients were retrospectively studied in whom imaging data including abdominal CT scan, MRI or Ultrasonography were routinely available. Available data on the mean steady state hematological/biochemical parameters (n=136),βs haplotypes(n=136), α globin gene status (n=105) and UGT1A1 genotypes (n=133) were reviewed from the respective medical records. Results: The mean serum total bilirubin level was significantly higher in the homozygous UGT1A1(AT)7 group as compared to UGT1A1(AT)6 group. Thus, not cholelithiasis but total serum bilirubin was influenced by UGT1A1 polymorphism in this SCA cohort. Conclusion: As observed in other population groups, the UGT1A1 (AT)7 homozygosity was significantly associated with raised serum total bilirubin level, but the prevalence of gallstones in the Omani SCA patients was not associated with α thalassaemia, UGT1A1 polymorphism, or βs haplotypes. |
Introduction
Chronic hemolysis in sickle cell anemia (SCA), results in
hyperbilirubinemia, as the water insoluble bilirubin needs to be
enzymatically converted into water soluble bilirubin glucoronides, for
its elimination through bile by uridine glucuronosyltransferase (UGT)
enzyme.[1,2] Chronic hyperbilirubinemia, thus over time, can lead to
the formation of gall stones (cholelithiasis), a common complication in
SCA.[1-3] Its onset can be as early as 2 to 4 years, but the prevalence
increases progressively with age.[4,5]
Inherited common sequence variations (polymorphisms) in the promoter region of the UGT1A1
gene that encodes the UGT enzyme had originally been associated with
Gilbert’s syndrome, a benign nonhemolytic hyperbilirubinemia in the
absence of liver disease.[6] The same variations had consistently been
associated, in various population groups, with hyperbilirubinemia in
several hemolysis-related clinical conditions viz. SCA, β thalassaemia and hereditary spherocytosis.[6-8]
These polymorphisms correspond to a simple sequence (TA) repeat number variation in the TATA promoter motif of UGT1A1
gene and had been shown to affect its expression. The alleles differ in
the number of repeats from 5 to 8 with (TA)[6] allele being the common
allele in Caucasians.[7] There is an inverse correlation between the
number of repeats and hepatic expression level of the UGT1A1 gene
on the one hand, and on the other, a direct correlation between the
number of repeats and bilirubinemia.[9,10] However, the relationship of
UGT1A1 polymorphism, both with
hyperbilirubinemia and with the incidence of cholelithiasis is not that
straightforward. Several inconsistencies raise the possibility that
other factors (genetic, environmental or both) may modulate either the
extent of hemolysis or the rate of gall stone formation or both.[10-12]
Studies of factors that could affect the hemolysis in SCA such
as α thalassaemia and HbF have also produced conflicting
data.[10-12] Other inconsistencies include the significantly lower
prevalence of cholelithiasis in African SCA patients as compared to
Jamaicans or African-Americans despite bearing similar African βs haplotypes.[1,9,10]
In
this regard, Haider et al., studying SCA patients from Kuwait, mostly
bearing the homozygous Arab-Indian haplotypes and high frequency of
alpha thalassaemia, report that the prevalence of gallstones was much
higher than that reported for Nigerian children with African βs
haplotypes. [13] This datum is intriguing given the known influence of
alpha thalassemia in reducing hemolysis. Such population and
geographical discrepancies in the incidence of cholelithiasis further
highlight the possibility that differences in the environmental
(dietary cholesterol/ fibers, use of third generation cephalosporins),
life style factors (fasting, smoking) and/or genetic factors other than
UGT1A1 may explain such
inconsistencies. Omani SCA patients offer an exceptional opportunity to
clarify some of the above mentioned issues, as both African and
Arab-Indian βs haplotypes in the
homozygous state are found in significant numbers sharing similar life
style and clinical interventional factors.[14] In this context, the
present single center cross sectional study allows certain homogeneity
in terms of clinical management/interventions.
This study investigates the influence of UGT1A1 polymorphism, HbF level, βs haplotypes and α thalassaemia on the steady state bilirubinemia and propensity to develop gall stones in Omani SCA patients.
Patients and Methods
The study patients were all from the hematology clinic at Sultan Qaboos University Hospital (SQUH). After getting approval from the hospital medical research and ethics committee and obtaining informed consent from patients or guardians, a total of 136 SCA patients were selected for whom imaging data (abdominal CT scan, MRI or Ultrasonography) performed as a routine study were available. (Figure1). Information on patients who underwent cholecystectomy was also recorded. The presence of sickle cell mutation was also confirmed at the DNA level. The βs-globin gene cluster haplotype, α globin gene status, and UGT1A1 polymorphism were determined as described earlier.[5,15-17] DNA sequencing of the polymerase chain reaction (PCR)-amplified entire β-globin gene segment (including the promoter, all exons, and exon-intron junctions) was performed on an ABI PRISM™ 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Data on the mean steady state hematological/biochemical parameters (n=136), βs haplotypes (n=136), α globin gene status (n=105) and UGT1A1 genotypes (n=133) were reviewed retrospectively from the medical records and utilized for this analysis. 61(44.85%) of these were on stable hydroxyurea (HU) therapy.
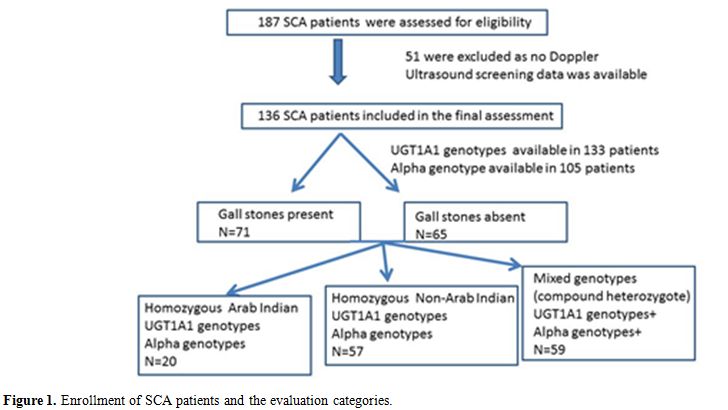 |
Figure 1. Enrollment of SCA patients and the evaluation categories. |
Statistical methods. Allele and genotype frequencies of the (TA)n
repeat were determined and tabulated. Deviations, if any, from the
expected Hardy-Weinberg equilibrium, was calculated by the Chi square
test. Differences in hematological data between different groups
(UGT1A1, α thalassaemia and Sickle haplotype groups) of patients
were assessed using Student’s t test and Chi square test. All the
analysis was performed using STATA ver. 11.1 (StataCorp, College
Station, TX, USA). A p value of <0.05 was considered as significant.
Results
Tables 1a and 1b summarize the relevant demographic (sex, age), red blood cell (Hb, HbF, reticulocytes), current therapy with HU and biochemical (total serum bilirubin) parameters along with the prevalence of gall stone for the 136 study patients for whom the status of cholelithiasis was available. Non statistically significant differences were noted between males and females in any of these parameters; respectively, Hb 9.7±1.6 vs. 9.1±0.9g/dl, HbF 8.9±6.7 vs. 10.1±6.6% and total serum bilirubin 50.9±36.7 vs. 42.2±44 µmol/L. None below ten years had stones, but the cumulative percentage of stones peaked in the fourth decade to 70.5%. The mean total serum bilirubin level reached the maximum in the second decade while the incidence of gall stones in the fourth decade. Overall 77 patients were homozygotes for βs haplotypes (20 - AI/AI & 57 - Ben/Ben or Ban/Ban) while all others were mixed haplotypes. In each decade age group, SCA patients with African haplotypes were more represented in percentage than those with the Arab-Indian haplotype.
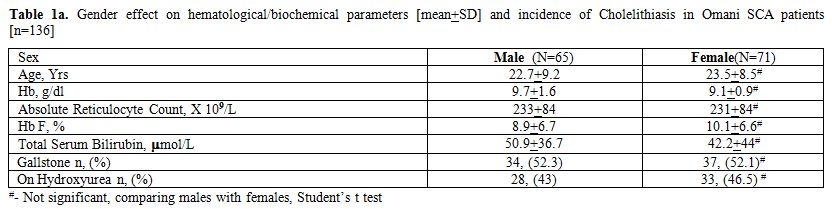 |
Table 1a. Gender effect on hematological/biochemical parameters [mean+SD] and incidence of Cholelithiasis in Omani SCA patients [n=136] |
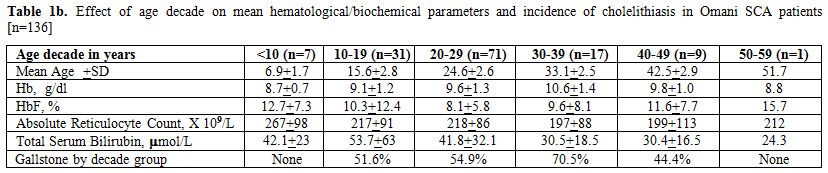 |
Table 1b. Effect of age decade on mean hematological/biochemical parameters and incidence of cholelithiasis in Omani SCA patients [n=136] |
In Table 2, the influence of the UGT1A1 promoter polymorphism, stratified into three groups as described by Chaar V et al[9] on the total serum bilirubin as well as on the prevalence of gall stones was examined along with the mean LDH, total Hb, HbF% and absolute reticulocyte count in 133 SCA patients. Two way comparisons show that the total serum bilirubin levels were significantly associated with UGT1A1 polymorphism but not with the prevalence of gall stones. No statistically significant difference was noted among the UGT1A1 genotype groups with respect to indicators of hemolysis (serum LDH, reticulocyte count, and Hb) and HbF level.
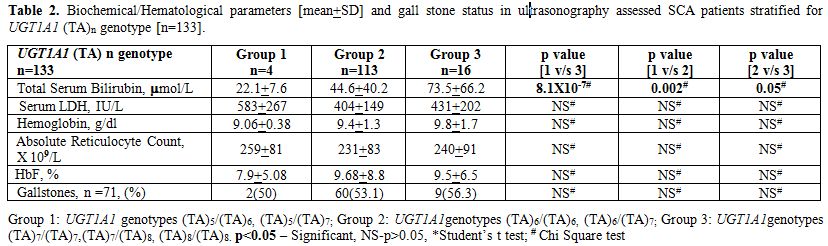 |
Table 2. Biochemical/Hematological parameters [mean+SD] and gall stone status in ultrasonography assessed SCA patients stratified for UGT1A1 (TA)n genotype [n=133]. |
Since α thalassaemia can modulate the rate of hemolysis, we examined its influence on these parameters. We first analyzed if the α globin genotypes were comparable between the UGT1A1 genotype groups. As expected and as shown in Table 3a, no difference was noted in the prevalence of α thalassaemia among the three UGT1A1 groups (n=105). As shown in Table 3b the homozygous state for αthalassaemia (-α/-α genotype) was significantly associated with all parameters examined (except the rate of gall stones) as compared to individuals with four alpha globin genes. Such difference persisted for total serum bilirubin and LDH in a two way comparison between subjects with –α/-α genotype and –α/αα genotype. The differences were restricted to total Hb and absolute reticulocyte count in the comparison between -α/αα and αα/αα genotypes.
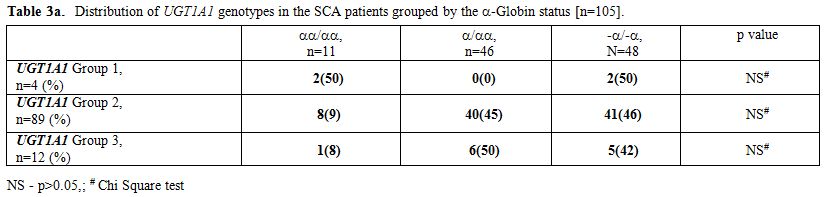 |
Table 3a. Distribution of UGT1A1 genotypes in the SCA patients grouped by the -Globin status [n=105]. |
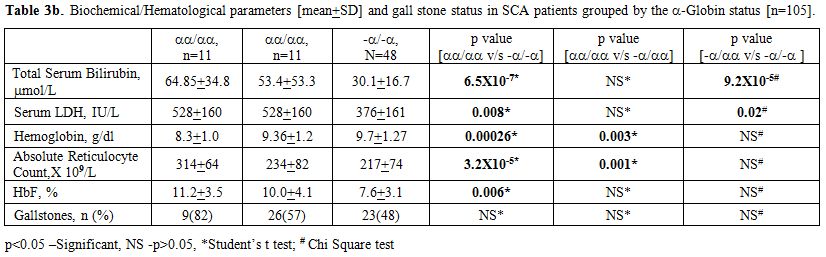 |
Table 3b. Biochemical/Hematological parameters [mean+SD] and gall stone status in SCA patients grouped by the -Globin status [n=105]. |
When a similar analysis was carried out for all these parameters based on homozygous βs
genotypes (Arab-Indian, Benin, and Bantu), we did not observe any
difference between the Benin and Bantu homozygotes (data not shown) but
both together were significantly different from the homozygotes for
Arab-Indian haplotype. This allowed us to combine both the Benin and
Bantu homozygotes into a single “African βs genotype” group for statistical comparisons against the Arab-Indian βs genotype.
First we checked for the alpha thalassemia status between the Arab-Indian and African βs genotype groups, and as shown in Table 4a, although overall prevalence of alpha thalassemic genotype was higher among African βs genotype group, the difference failed to reach statistical significance (p>0.05, chi square test). Data in Table 4b show that the subjects homozygous for the Arab-Indian βs genotype are distinct from those having the African βs
genotypes in terms of bilirubin and hemolysis-related factors (lower
total serum bilirubin, serum LDH and absolute reticulocyte count but a
higher Hb and HbF%).
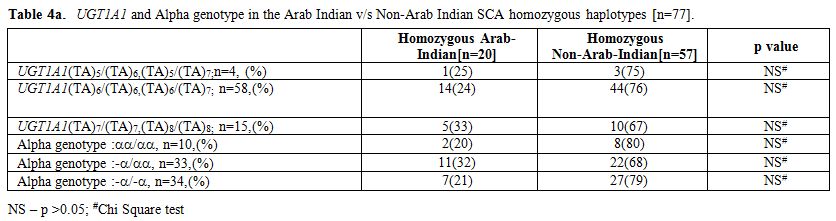 |
Table 4a. UGT1A1 and Alpha genotype in the Arab Indian v/s Non-Arab Indian SCA homozygous haplotypes [n=77]. |
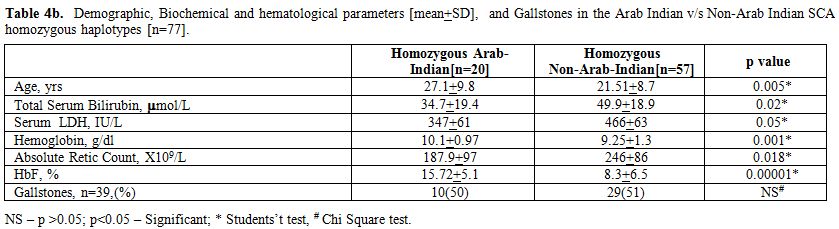 |
Table 4b. Demographic, Biochemical and hematological parameters [mean+SD], and Gallstones in the Arab Indian v/s Non-Arab Indian SCA homozygous haplotypes [n=77]. |
Discussion
Acknowledgements
We wish to thank the Hospital Administration for allowing the use of hospital data. This work was supported in part by a grant (RC/MED/HAEM/10/01) from the “The Research Council” of Oman.
References

[TOP]