Received: August 23, 2015
Accepted: November 18, 2015
Mediterr J Hematol Infect Dis 2016, 8(1): e2016001, DOI 10.4084/MJHID.2016.001
This article is available on PDF format at:

| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Introduction. In males,
acquired hypogonadotropic hypogonadism (AHH) includes all disorders
that damage or alter the function of gonadotropin-releasing hormone
(GnRH) neurons and/or pituitary gonadotroph cells. The clinical
characteristics of AHH are androgen deficiency and lack, delay or halt
of pubertal sexual maturation. AHH lead to decreased libido, impaired
erectile function, and strength, a worsened sense of well-being and
degraded quality of life (QOL). Patients and methods. We studied 11 adult men with thalassemia major (TM) aged between 26 to 54 years (mean ± SD: 34.3 ± 8.8 years) with AHH. Twelve age- and sex-matched TM patients with normal pubertal development were used as a control group. All patients were on regular transfusions and iron chelation therapy. Fasting venous blood samples were collected two weeks after transfusion to measure serum concentrations of IGF-1, free thyroxine (FT4), thyrotropin (TSH), cortisol, luteinizing hormone (LH), follicle stimulating hormone (FSH), total testosterone (TT), prolactin and estradiol (E2), glucose, urea, creatinine and electrolytes (including calcium and phosphate). Liver functions and screening for hepatitis C virus seropositivity (HCVab and HCV-RNA) were performed. Iron status was assessed by measuring serum ferritin levels, and evaluation of iron concentrations in the liver (LIC) and heart using MRI- T2*. Bone mineral density was measured at the lumbar spine (L1-L4) for all patients with AHH by dual energy X-ray absorptiometry (DXA) using Hologic QDR 4000 machine. Results. The mean basal serum LH and FSH concentrations in AHH patients were 2.4 ± 2.2 IU/L and 1.2 ± 0.9 IU/L respectively; these, values were significantly lower compared to the control group. Semen analysis in 5 patients with AHH showed azoospermia in 3 and oligoasthenozoospermia in 2. The percentage of patients with serum ferritin level >2000 ng/ml (severe iron load) was significantly higher in AHH patients compared to controls, 5/11 (45.4 %) versus 1/12 (8.3%), p=0.043. Heart iron concentrations (T2* values) were significantly lower in AHH patients compared to controls (p=0.004). Magnetic resonance imaging in the 3 azoospermic patients revealed volume loss and reduction of pituitary signal intensity. Heart T2* values were significantly reduced in the AHH group vs. the controls (p=0.004). On the other hand, liver iron concentration (mg/g dry weight) was not different between the two groups of TM patients. Using DXA, 63.6 % (7/11) of patients with AHH were osteoporotic, and 36.3 % (4/11) were osteopenic. Conclusions. In this cohort of thalassemic patients iron overload and chronic liver disease appear to play a role in the development of AHH. Treatment of AHH in TM patients is a vital and dynamic field for improving their health and QOL. Early identification and management of AHH are very crucial to avoid long-term morbidity, including sexual dysfunction and infertility. Therapy aims to restore serum testosterone levels to the mid–normal range. Many exciting opportunities remain for further research and therapeutic development. |
Introduction
In males, acquired hypogonadotropic hypogonadism (AHH) includes all
postnatal disorders that damage or alter the function of
gonadotropin-releasing hormone (GnRH) neurons and/or pituitary
gonadotroph cells. The anatomical, histological and functional changes
of AHH encompass a vast range of causes including infiltrative and
infectious pituitary lesions (e.g., hemosiderosis, sarcoidosis and
histiocytosis), hyperprolactinemia, brain injury and pituitary
irradiation.[1-4]
In adolescents, the clinical
presentations of AHH include lack, delay and/or halt of pubertal sexual
maturation. Adults with AHH have decreased libido, erectile
dysfunction, worsened sense of well-being and lower quality of life
(QOL). Physical examination is usually normal in hypogonadism of
recent-onset. In long-standing AHH, diminished facial and body hair
decreased muscle mass, and appearance of fine facial wrinkles,
gynecomastia, and small testes are observed. Spermatogenesis is
impaired, and the volume of ejaculate is decreased only when
gonadotropins and testosterone levels are very low. Low serum
concentrations of testosterone and gonadotropins confirm the diagnosis
of AHH.[1-7]
Adult male patients with TM, on
frequent blood transfusions, are predisposed to develop AHH. The
prevalence of AHH in TM depends mainly on the age composition of the
thalassemia cohort and the degree of compliance with blood transfusion
and mainly chelation programs.
We report 11 adult men with
thalassemia major (TM) and AHH. They presented with loss of libido
and/or infertility; reduced shaving frequency; erectile or ejaculatory
dysfunction; diagnosis of AHH was confirmed by appropriate laboratory
tests.[8,9] They were selected from a cohort of adult
TM patients with significant compliance to blood transfusion and
efficient implementation of chelation programs.
Patients and Methods
Setting and study design:
The study started at the beginning of 2009 by VDS, (Coordinator of
International Network of Clinicians for Endocrinopathies in Thalassemia
and Adolescent Medicine ICET-A) at the Thalassaemia Centre of Ferrara[10]
and was completed by the end of August 2015 at the Quisisana Pediatric
and Adolescent Outpatient Clinic of Ferrara. During this period, 11
adults with TM (aged between 26 to 54 years, mean ± SD: 34.3 ± 8.8
years) were diagnosed with hypogonadotropic hypogonadism and were
studied.
All patients had spontaneous, full pubertal development.
At presentation, patients complained of sexual dysfunction and/or
fertility problems, loss of libido or infertility, and erectile or
ejaculatory dysfunction (diminished erectile quality and frequency;
diminished early morning erections; decreased or watery semen
production). There was no history of anosmia, cryptorchidism, brain
injury, exposure to chemicals or drug abuse. Patients had no family
history of infertility, mental, physical or pubertal development
retardation.
A randomly selected 12 age- and sex-matched TM
patients with spontaneous and full pubertal development, who visited
our outpatient clinics for clinical and laboratory endocrine assessment
served as controls.
Ethical approval for the study was obtained
in accordance with local institutional requirements in accordance with
the Declaration of Helsinki (http://www.wma.net). All procedures were
carried out with the adequate understanding and consent of patients.
Exclusion criteria:
Exclusion criteria included: 1) treatment with androgens or anabolic
steroids within the last 3 months; 2) hyperprolactinemia or treatment
with prolactin-lowering drugs; 3) mental illness (depression, anxiety
disorders, eating disorders and addictive behaviors). 4) acute
diseases; 5) accidental severe head trauma and brain injury; 6)
alterations in nutritional status with significant loss of weight
and/or the presence of depression; 7) smoking of more than 15
cigarettes/day or alcohol abuse (more than three glasses of wine/day);
and 8) increased estrogen levels secondary to liver disease.
Research design:
Extensive history was taken including data on associated complications
of thalassemia and current medications. Thorough physical examination
was completed including anthropometry (weight, height, BMI), vital
signs (blood pressure, heart rate) and genital status. Body mass index
(BMI) was calculated (weight in Kg/ height in m2). A subject was considered overweight when the BMI was between 25 and 30 and obese above 30.[11]
Age at first transfusion, interval between transfusions, type, and
compliance to iron chelation and associated endocrine complications
were recorded. [11]
Blood sampling and analytical procedures:
All blood samples were collected in the morning (08.00 – 09.00 am),
after an overnight fast, and 2 weeks after blood transfusion. The
circulating concentrations of IGF-1, free thyroxine (FT4), thyrotropin
(TSH), cortisol, luteinizing hormone (LH), follicle stimulating hormone
(FSH), total testosterone (TT), prolactin, estradiol (E2), glucose,
urea, creatinine and electrolytes (including calcium and phosphate)
were measured.
To exclude severe liver injury and dysfunction,
serum concentrations of alanine aminotransferase (ALT), gamma glutamyl
transferase (γ GT), alkaline phosphatase (ALP), total and direct
bilirubin, total proteins, albumin, prothrombin time (PT) and
international normalization ratio (INR) were measured. Screening assays
for hepatitis C virus seropositivity (HCVab and HCV-RNA) were performed
applying routine laboratory methods.
Plasma total IGF-1 was
measured by a chemiluminescent immunometric assay (CLIA) method
(Nichols Institute Diagnostics, San Juan, CA). The assay was performed
after separation of IGF-1 from binding proteins by Liaison®
autoanalyzer (DiaSorin SpA, Saluggia, Italy). A detailed description of
the method and the evaluation of the results have been recently
published.[12] The sensitivity of the test was 6
ng/ml, whereas the intra- and interassay coefficients of variation
(CVs) of our in-house pooled serum control sample were 4.8% and 6.7%,
respectively. The reported analytical sensitivity of this assay was
from 6 to 25 ng/ml (normal values set at the 2.5th-97.5th percentile were: 95.6-366.7 ng/ml for ages 25 to 39 yrs, 60.8-297.7 ng/ml for 40 to 59 yrs).[12]
The
other hormonal, biochemical and hematogical parameters were determined
using commercially available automated chemiluminescence immunoassay
and other systems. The intra- and interassay CV for all methods were
< 5.8% and < 7.8%, respectively.
Definitions of endocrine disorders: Low blood testosterone levels and low pituitary gonadotropin levels (LH and FSH) indicated a diagnosis of HH.[7,9,11] Diabetes and secondary TSH deficiency were defined as previously described.[11] Adrenal insufficiency was diagnosed if basal cortisol was 3.5 μg/dl (98 nmol/liter) or less.[13] Hyperprolactinemia was defined as a basal level greater than the locally derived normal assay reference range.
Assessment of iron overload:
Iron overload was assessed by direct and indirect methods. At the
beginning of the study, it was evaluated by measuring serum ferritin
level. Iron status was classified as mild (ferritin < 1000 ng/ml),
moderate (ferritin >1000 ng/ml and < 2000 ng/ml) or severe
(ferritin >2000 ng/ml).[14] Commercial reagents were used for the determination of serum ferritin levels based on an immune- enzymatic method.
In
6 TM patients with AHH and 8 patients without AHH, heart iron was
assessed by Magnetic resonance imaging (MRI) using a 1.5 T scanner (GE
Signa/Excite HD, Milwaukee, WI, USA) within the Myocardial Iron
Overload in Thalassemia (MIOT) network, where MRI scans are performed
using homogeneous, standardized and validated procedures.[14,15] A traditional cut-off value of heart T2* > 20 ms was considered normal.[15] In the same patients, liver iron concentration (LIC) was assayed by MRI.[16] Liver T2* values were converted into MRI liver iron content (LIC) values sing the calibration curve introduced by Wood et al.[18]
In two TM patients with AHH and one patient without AHH, LIC was
quantified by biopsy using atomic absorption spectrophotometry or
Superconducting Quantum Interference Device (SQUID) and the LIC values
were expressed as mg/g dry weight (dw).[18] LIC (mg Fe/gr dw) was classified as mild (LIC > 3 and < 7), moderate (LIC > 7 and < 14) and severe (LIC > 14).[14]
Bone
mineral density (BMD) at the lumbar spine (L1-L4) was assessed for all
AHH patients using dual energy x-ray absorptiometry (DXA) by Hologic
QDR 4000 machine (Bedford, MA).
Osteopenia or osteoporosis was
defined according to World Health Organization (WHO) criteria, based on
BMD expressed as Z-score: osteopenia (Z-score between –1 to –2.5 SD)
and osteoporosis (Z-score < –2.5 SD).[19]
Z-score is the number of standard deviations above or below what is
normally expected for someone with the same age, sex, and ethnic
origin. Osteoporosis is a common disorder of reduced bone strength that
predisposes to an increased risk for fractures in older individuals.
Three
TM patients with AHH underwent pituitary MRI using a 1.5T scanner
(Sonata, Siemens Medical, Erlanger, Germany). Pituitary-to-fat signal
intensity ratios (SIR) were calculated from coronal T2-weighted images.
Estimated pituitary volumes were measured using pituitary height,
width, and length on T1-weighted images.
Statistical analysis.
Standard computer program SPSS for Windows, release 13.0 (SPSS Inc,
Tulsa, IL, USA) was used for data entry and analysis. All numeric
variables were expressed as mean ± standard deviation (SD).
Comparison of different variables in the two groups was done using
unpaired - student t-test and Mann-Whitney test for normal and
nonparametric variables respectively. Chi-square (X2)
test was used to compare the frequency of qualitative variables among
the different groups. Pearson’s and Spearman’s correlation tests were
used to study correlations between variables with parametric and
non-parametric distributions respectively. p < 0.05 was considered
significant.
Results
Patients’ characteristics:
All patients were on regular transfusions (pre-transfusional hemoglobin
level 9 ± 0.3 g/dl) but different iron chelation regimes therapy. Ten
patients were on deferoxamine (DFO) 30-45 mg/kg body weight, 4-6 days a
week by slow subcutaneous infusion by the pump. Nine patients were on
oral deferiprone (DFP) 75 mg/kg body weight daily. Two patients were on
combined deferiprone plus deferoxamine 75 mg/kg body weight daily and
40 mg/kg body weight, 3 days a week, by slow subcutaneous infusion and
2 on oral deferasirox (DFX) 25-30 mg/kg body weight daily. Chelation
therapy has been changed over time. Treatment with intramuscular DFO
was available since 1969, DFP since 1995 and DFX since 2007.
The baseline demographic and clinical data of the AHH and non-AHH groups of adult patients with TM are summarized in Table 1.
The mean (± SD) age and BMI did not differ between TM patients with and
controls (without) AHH. All AHH patients had spontaneous, and full
pubertal development, and 2 reported previous paternity.
The
presenting symptoms in patients with AHH included: loss of libido (7
patients), infertility (2 married patients); diminished erectile
quality and frequency, diminished early morning erections (9 patients);
fatigue (1 patient); ejaculatory dysfunction, decreased or watery semen
production (11 patients). Symptoms were present for 2 – 9 months before
endocrine evaluation; and were attributed to the “iron overload,
chronic diseases itself, low hemoglobin level, liver dysfunction,
associated endocrine complications (diabetes, hypothyroidism)”.
Patients had no history of anosmia, cryptorchidism, head injury,
exposure to chemicals, or alcohol and drug abuse. They had no family
history of infertility, or mental, physical or pubertal development
retardation.
The demographic and chelation data, the co-existent
endocrinopathies and the biochemical, hormonal and iron-load results of
patients with AHH and controls are summarized in tables 1 and 2.
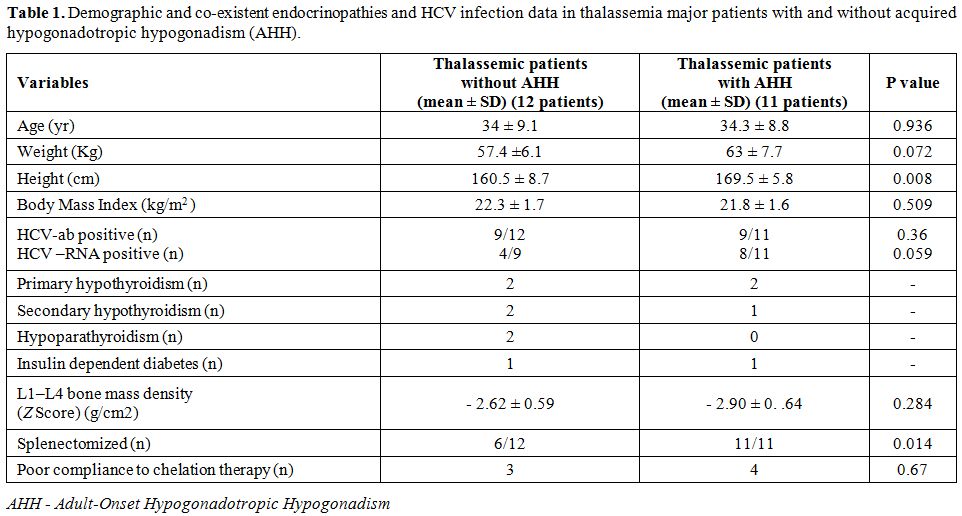 |
Table 1. Demographic and co-existent endocrinopathies and HCV infection data in thalassemia major patients with and without acquired hypogonadotropic hypogonadism (AHH). |
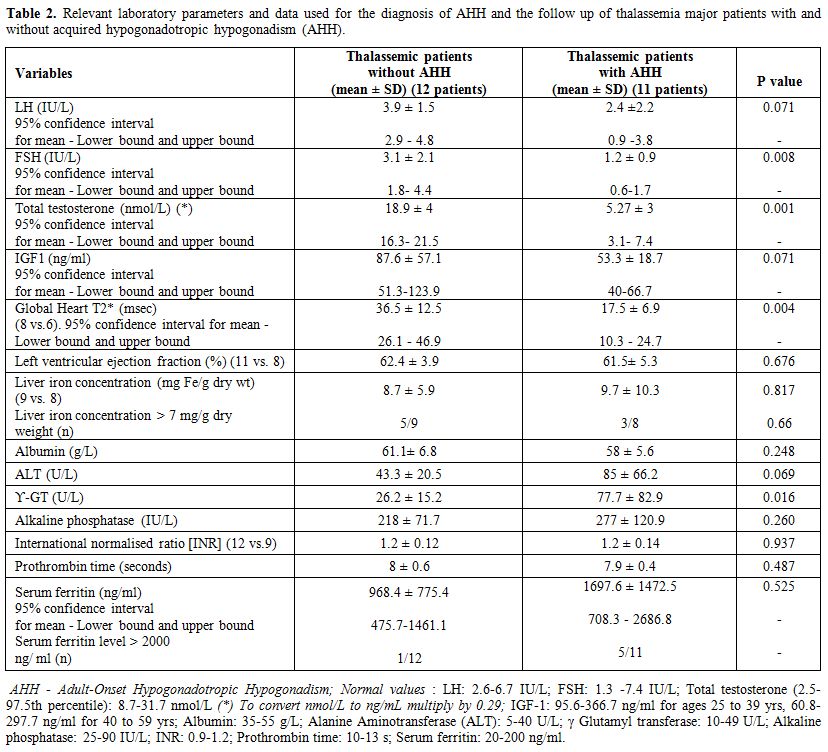 |
Table 2. Relevant laboratory parameters and data used for the diagnosis of AHH and the follow up of thalassemia major patients with and without acquired hypogonadotropic hypogonadism (AHH). |
TM patients with AHH were taller compared to non-AHH patients (169.5 ± 5.8 cm vs. 160.5 ± 8.7 cm; p: 0.008) (Table 1).
In all patients, genital examination revealed a normal adult-sized
penis and testis volume between 12–25 mL. All patients with AHH had
undergone splenectomy due to hypersplenism and/or massive splenomegaly,
versus 50% of the control group.
The mean baseline serum
luteinizing hormone and follicle-stimulating hormone concentrations in
AHH patients were 2.4 ± 2.2 IU/L (95% confidence interval for mean: 0.9
-3.8 IU/L) and 1.2 ± 0.9 IU/L (95% confidence interval for mean:
0.6-1.7 IU/L), respectively. These values were significantly lower
compared to controls. The circulating concentrations of TT and IGF-I
were significantly lower in patients with AHH group versus controls. In
all patients with AHH plasma IGF- 1 concentrations were below the 2.5th
percentile of normal Italian population of the same age range. Growth
hormone after stimulation test was not assessed. In patients with AHH
serum estradiol and prolactin concentrations were normal. 58.3% of
patients with AHH and 36.3% of controls had other associated
endocrinopathies. (Table 1). None had a basal cortisol equal or below 3.5 μg/dl (98 nmol/liter).
Five
patients with AHH provided semen specimens for testing. Three had
azoospermia, and two had severe oligoasthenozoospermia. Magnetic
resonance imaging in three azoospermic patients revealed pituitary
volume loss and a reduction in signal intensity.
Osteoporosis
was diagnosed in 63.6 % (7/11) and osteopenia in 36.3 % (4/11) of the
TM patients with AHH using DXA scan (Table 1).
Patients
with AHH had significantly higher serum γGT concentrations and
non-significantly higher ALT concentrations compared to controls.
HCV-RNA seropositive were 8/11(72%) of patients with AHH versus 4/9
(44%) of controls (p=0.059) (Table 1). Serum ferritin, albumin, and
alkaline phosphatase concentrations did not differ between the two
groups (Table 2).
Assessment of iron overload:
Serum ferritin level >2000 ng/ml (severe iron overload) was present
in 5 patients (45.4 %) with AHH versus 1 patient (8.3%) without AHH (Table 2).
Heart T2* values were significantly reduced in the AHH group vs.
controls (p=0.004). On the other hand, liver iron concentration (mg/g
dry weight) was not different between the two groups.
Other correlations:
A significant correlation was found between FSH and total testosterone
(r = 0.632, p=0.002); total testosterone and IGF1 (r = 0.590, p=0.005)
and IGF-1 and ϒGT (r = - 0.422, p=0.050). Total testosterone was
correlated significantly with the height (r = - 0.552, p=0.008). No
correlation was observed between serum ferritin on the one hand and
serum LH, FSH, and TT concentrations on the other hand.
Discussion
Hypogonadotropic hypogonadism (HH) is the most frequent endocrinopathy in transfused patients with TM.[11,20]
Patients with TM usually suffer from iron overload as a consequence of
frequent transfusions and ineffective erythropoiesis. Iron has a
catalytic role that produces powerful reactive oxidant species (ROS)
and free radicals, which leads to oxidative damage.[21]
The sensitivity of different organs to accumulate iron varies
considerably. Iron accumulates in tissues with high levels of
transferrin receptor such as liver, heart and endocrine glands. The
anterior pituitary gland is particularly sensitive to free radical
oxidative stress that may impair gonadotropins and growth hormone (GH)
secretion. Consequently thalassemic patients with marked hemosiderosis
are predisposed to develop hypogonadism and short stature.[20]
The
best predictor of pituitary iron overload is the detection of decreased
signal intensity of the anterior lobe of the pituitary gland on
T2-weighted MRI.22 Unfortunately at the time of the study this
technique was available only for the three azoospermic TM patients with
AHH.
Our patients with AHH had significantly elevated levels of γ
GT and higher prevalence of HCV-RNA seropositivity and associated
endocrinopathies compared to controls without AHH. These findings point
to the importance of liver disease and associated endocrine disorders
as important contributing factors in the etiology of HH.[11,20]
The
study also strongly supports that AHH in TM patients is associated with
a reduced heart T2* (ms). Myocardial T2* values < 20ms (1.1 mg/g.dw)
indicate cardiac iron overload. The vast majority of patients who
present with heart failure caused by cardiac iron overload have T2*
< 10 ms and low T2* values are powerful predictors of the subsequent
development of cardiac failure. Therefore early detection of low
myocardial T2* (< 20 ms) is important for an early treatment of
cardiac iron overload and prevention of cardiac failure.[16,17,22]
Recently,
it was shown that blood transfusion produces significant acute changes
in the hormonal milieu and sperm parameters of patients with iron
overload.[23,24] Moderate hypoxia has been shown to decrease gonadotropin secretion within 2 days of arrival at moderate altitude.[25]
Our
TM patients with AHH had significantly lower serum LH, FSH,
testosterone concentrations and higher γ GT levels compared with
control patients without AHH. Moreover, they showed significantly
higher levels of hepatic iron overload and higher prevalence of serum
ferritin level > 2000 ng/ml. This abnormal iron overload status
similarly involves the pituitary gland and causes progressive,
irreversible damage to LH, FSH, and GH secretion. However, a potential
negative effect of chronic intermittent anemia on gonadotropins
secretion cannot be excluded, even though our patients were on regular
transfusion regimens (pre- transfusional Hb level 9.0 ± 0.3 g/dl). A
pre-transfusion Hb level of 9 g/dl may not be capable of suppressing
adequately bone marrow (BM) activity and intestinal absorption of iron.
In support of this view, all patients with AHH had undergone
splenectomy due to hypersplenism and/or massive splenomegaly, versus
50% of the control group. This difference implied that transfusion
therapy was less efficient in suppressing BM hyperactivity and
extramedullary hemopoiesis that leads to marked splenic enlargement.
An
accurate assessment of the prevalence rate of AHH in adults with TM is
difficult, and under-diagnosis is common. There are few data in the
literature studying this new emerging complication. The reported
prevalence varies from 8.3% to 12%.[26-28] Albu et
al. reported that TM patients with AHH were significantly older (median
age 26 vs. 16.5 years, p: 0.007) and had higher serum ferritin levels
compared to patients without AHH.[26] TM patients
rarely consult doctors due to a lack of obvious and evident symptoms of
AHH. In our TM patients with AHH the presenting symptoms were a loss of
libido (7 patients) or infertility (2 married patients); incomplete
and/or not persistent erection (9 patients); fatigue (1 patient) and
ejaculatory dysfunction (decreased or watery semen production: 11
patients). These symptoms had been present for a mean of 6 ± 2 months
(range 2 – 9 months) before the first endocrine evaluation. They were
previously attributed to the chronic disease itself, iron overload, low
hemoglobin level, liver dysfunction, associated endocrine complications.
Early
identification and management of AHH are very crucial to avoid
subsequent long-term morbidity, including infertility, sexual
dysfunction, osteoporosis, weakness and disturbed QOL.[1-4]
The
concentration of serum TT reaches its maximum around 25–30 years of age
and starts a slow, steady decline thereafter at a rate of about 1% per
year.[8] Furthermore, there is a 1.2% annual increase
in sexual hormone-binding globulin (SHBG), which makes it unavailable
to the tissues.[29]
In men, 60% of circulating
testosterone is bound to sex hormone- binding globulin (SHBG), 38% is
bound to albumin, and only 2% is unbound or free. Total testosterone
levels might be normal with hypogonadism if the SHBG levels are
increased. Levels of SHBG increase with age, causing a decrease in
bioavailable testosterone.[1-4] SHBG levels are
elevated in patients with cirrhosis due to increased hepatic
production, but the pathogenesis of this remains not fully explained.
Rising levels of SHBG have been shown to correlate also with severity
of fibrosis in patients with the chronic liver disease.[7]
If testosterone levels are low-normal but the clinical symptoms and
signs indicate hypogonadism, measurement of serum total testosterone
levels should be repeated and an SHBG level should be determined, and
the bioavailable testosterone levels can be calculated.
Symptoms
of androgen deficiency need to be specifically inquired about if
hypogonadism is suspected, although none of these symptoms are specific
to the low androgen state. Questionnaires such as Aging Male Symptom
Score (AMS) and Androgen Deficiency in Aging Men (ADAM) are not
recommended for the diagnosis of hypogonadism because of low
specificity.[30-33]
Hormone replacement therapy
can significantly improve the QOL of patients by restoring sexual
function. The cut-off values to diagnose hypogonadism have been
variable. Recently an American consensus statement reported that above
11.1 nmol/L TT is normal, below 6.9 nmol/L is diagnostic of
hypogonadism, and 6.9–11.1 nmol/L is equivocal.[34] In Europe, those figures are slightly different (12, 8 and 8–12 nmol/L respectively).[35]
The diagnosis of hypogonadism should never be based on a single
testosterone level. For those patients with low testosterone, repeated
measurement of testosterone level at least 1 month apart, is necessary
to confirm a low testosterone level. Those with a total testosterone
level between 8 –12 nmol/l are classified as borderline. In borderline
cases, the SHBG level can be measured to calculate the free
testosterone using a mathematical formula (www.issam.ch),
or the free testosterone can be measured using the equilibrium dialysis
method. However, if further assays are not available, and patients are
symptomatic, a trial of testosterone therapy can be given followed by
re-assessment after three months. Those with a level above 12 nmol/l
are unlikely to be hypogonadal and should not receive testosterone
treatment.[8]
It is important to differentiate
adult-onset HH, (characterized by frankly low serum testosterone levels
in the presence of low or normal gonadotropins) from the progressive
testosterone deficiency observed in a small minority of aging men,
known as late-onset hypogonadism (LOH). This latter condition has been
defined as a syndrome in middle-aged and elderly men reporting sexual
symptoms in the presence of moderately low total testosterone levels,
with variable levels of gonadotropins, which mostly involves gonadal
components in its pathogenesis.[36]
The choice
of therapy in males with AHH depends on the fertility requirements of
the patients. When fertility is desired, gonadotropin therapy is
necessary to induce spermatogenesis.[36] Different
treatment protocols can be used. The typical gonadotropin regimen
combines human chorionic gonadotropin (hCG) and FSH.[37,38]
Two of our azoospermic TM patients received a combination of hCG and
hFSH that resulted in spermatogenesis (oligoasthenozoospermia) within 6
months.
Testosterone replacement is another convenient therapy if
fertility is not in question. Testosterone replacement is recommended
for symptomatic classical androgen deficiency syndromes after excluding
contraindications in the initial work up. Androgen deficiency can be
treated using any one of the approved testosterone formulations after
consideration of pharmacokinetics, patient preference, cost, and
potential formulation-specific adverse effects. Adverse events are
reduced high-density lipoprotein cholesterol, increased prostatic
symptoms and increased cardiovascular risk.[39] Therefore, testosterone therapy should be accompanied by a standardized monitoring plan and general health evaluation.
This
study was limited by incomplete data on statural growth in thalassemia
major parents. Potential causative factors of shorter final height in
TM patients without AHH compared to patients with AHH include genetic
factors, previous severe iron overload, desferrioxamine “toxicity”,
delayed puberty and defects in the growth hormone-insulin- like growth
factor-1 (GH-IGF-1) axis.
A second limitation is that our study
did not localize the anatomic level of HPG axis dysfunction. Although a
strong association between pituitary R2 and pituitary volume with
clinical disease suggests that secondary hypogonadism is the dominant
etiology, we cannot exclude tertiary hypogonadism. Further, targeted
studies are needed to address these questions and to explore the
potential metabolic syndrome related to hypogonadism in our TM patients.
Conclusions
In adult eugonadal thalassaemic patients, annual screening for the
development of hypogonadism should be performed. This should include
history (libido, erectile function, the frequency of spontaneous
erections), physical examination and biochemical assessment (SHBG and
serum fasting testosterone in the early morning).
In our
thalassemic patients iron overload and chronic liver disease appear to
play a role in the development of AHH. Studying TM patients with AHH
has become a vital and dynamic field for improving their health and
QOL. Many exciting opportunities remain for further research and
therapeutic development. Treatment is in the form of testosterone
replacement therapy in a variety of preparations. Therapy aims to
restore serum testosterone to the mid–normal range and correct symptoms
and signs of androgen deficiency. However, the results and safety of
long-term prospective controlled trials of testosterone therapy are
still awaited.
References

[TOP]