Received: February 15, 2016
Accepted: March 23, 2016
Mediterr J Hematol Infect Dis 2016, 8(1): e2016023, DOI 10.4084/MJHID.2016.023
This article is available on PDF format at:

Marcus Kühn, Kety Sammartin, Mitja Nabergoj and Fabrizio Vianello
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Peripheral neuropathy is a common
complication of arsenic toxicity. Symptoms are usually mild and
reversible following discontinuation of treatment. A more severe
chronic sensorimotor polyneuropathy characterized by distal axonal-loss
neuropathy can be seen in chronic arsenic exposure. The clinical course
of arsenic neurotoxicity in patients with coexistence of thiamine
deficiency is only anecdotally known but this association may
potentially lead to severe consequences. We describe a case of acute irreversible axonal neuropathy in a patient with hidden thiamine deficiency who was treated with a short course of arsenic trioxide for acute promyelocytic leukemia. Thiamine replacement therapy and arsenic trioxide discontinuation were not followed by neurological recovery and severe polyneuropathy persisted at 12-month follow-up. Thiamine plasma levels should be measured in patients who are candidate to arsenic trioxide therapy. Prophylactic administration of vitamin B1 may be advisable. The appearance of polyneuropathy signs early during the administration of arsenic trioxide should prompt electrodiagnostic testing to rule out a pattern of axonal neuropathy which would need immediate discontinuation of arsenic trioxide. |
Case Report
Although polyneuropathy is common following arsenic trioxide therapy
of subjects with acute promyelocytic leukemia (APL), symptoms are
generally mild and they disappear after completion of arsenic
treatment. In November 2014, a 72-year old Caucasian woman was admitted
to this Unit with fever and pancytopenia (white blood cell count 0.19x109/L, hemoglobin 76 g/L, platelets 41x109/L).
She had a medical history of lobular carcinoma of the left breast
treated with mastectomy and chemotherapy in 2012, Hashimoto
thyroiditis, arterial hypertension. There was no prior neurological
disorder and physical examination was unremarkable. Coagulation tests
showed reduced prothrombin time ratio (59%) and normal activated
partial thromboplastin time, a slightly reduced fibrinogen (1,4 g/L)
and increased D-dimer (8634 µg/L). Bone marrow evaluation was
consistent with acute promyelocytic leukemia (APL) according to
French-American-British (FAB) classification system. The patient was
started on all-trans retinoic acid (45 mg/m2)
according to protocol APL0406[1] and, by day 3, arsenic trioxide (ATO)
10 mg/d was added at the time of PML/RAR alpha rearrangement
identification by PCR. The clinical course was complicated by
differentiation syndrome, which manifested as mild increase of
creatinine levels, peripheral edema and pleuro-pericardial effusion.
All-trans retinoic acid was therefore stopped on day 12, dexamethasone
and furosemide added and the patient was continued on arsenic trioxide.
On
day 16, the patient gradually regained kidney function and all-trans
retinoic was resumed. Starting on day 22, progressive cognitive
impairment, insomnia, slurred speech, spatial and temporal
disorientation occurred. As this worsening clinical picture was
concomitant to hypernatremia (up to 160 mmol/L, reference values
136-145 mmol/L), neurological symptoms were attributed to
sodium-related hyperosmolarity.
Over the following days an
impaired level of consciousness and lethargy was noticed. Progressive
and appropriate normalization of natriemia did not improve neurological
dysfunction.
Additional diagnostic procedures were performed. A
CT scan of the brain did not reveal abnormalities related to the acute
clinical picture, showing only age-related mild cerebral and cerebellar
atrophy. Cerebrospinal fluid showed only mild protein
elevation.
Plasma
levels of vitamin B1 were low (22 nmol/L, reference values 66-200
nmol/L) and supplementation was started. Over the following 5 days the
patient showed a slow but progressive full recovery of cognitive
functions.
Unexpectedly, at normalization of cognitive function,
a pattern of symmetric peripheral neuropathy involving upper and lower
limbs became clinically evident. Physical examination showed that left
and right hands were severely weak, particularly the abductor pollicis
brevis, with mild strength reduction in the proximal arms. Deep-tendon
reflexes were absent. There was bilateral foot drop, and the patient
was unable to walk. Strength in the ankle dorsiflexors, extensor
hallucis longus, extensor digitorum brevis, and toe flexors was
severely compromised. There was no muscle atrophy or fasciculation, and
the remainder of the neurologic examination was normal.
Electromyography (EMG) showed a severe sensory-motor axonal neuropathy
of the upper and lower extremities with a greater reduction of sensory
action potential.
Meanwhile on day 28 arsenic trioxide had been
stopped according to the therapy protocol. Bone marrow evaluation
showed molecular remission. At that time the patient was bedridden
because of a severe neurological impairment, so we chose not to
administer consolidation therapy as it would have been required
according to standard protocol.[1] The patient was then continued on
maintenance therapy with methotrexate and 6-mercaptopurine. Molecular
remission was confirmed in bone marrow aspirate at the 13-month
follow-up.
Over the following on 6 months from arsenic trioxide
discontinuation, the patient regained full proximal muscle strength but
with almost complete persistence of marked distal weakness of the
hands, ly finger and wrist extensors, weakness of foot dorsiflexors
bilaterally and the inability to maintain an upright position.
Neurological examination and EMG were substantially unchanged at the
12-month follow-up evaluation.
We believe that occult thiamine
deficiency exacerbated arsenic trioxide neurotoxicity causing an
unexpected and irreversible distal axonal symmetric neuropathy. This is
the first report of acute and irreversible axonal neuropathy in a
patient treated with arsenic trioxide with a background of thiamine
deficiency. Peripheral neuropathy has long since been associated with
the use of arsenic. Several clinical studies have been published on ATO
treatment of patients with APL (Table).[2-12]
Although details on grade and duration of peripheral neuropathy were
not always clearly included, overall grade 3/4 neuropathy accounts for
about 0.5% of patients treated with ATO and recovery is always
observed. Yip et al reported a case of severe neurotoxicity during
arsenic therapy in a subject with APL and occult thiamine
deficiency.[13] However, thiamine administration led to rapid
improvement suggesting a major role of thiamine deficiency over arsenic
toxicity.
Severe and irreversible arsenic neurotoxicity in the
setting of thiamine deficiency can be explained by their common
metabolic target. In fact, thiamine deficiency and arsenic exposure
severely impair pyruvate dehydrogenase (PDH) activity, an enzyme
responsible for converting glucose to energy in high yield. As neural
tissues are highly dependent on carbohydrates for energy production and
cellular metabolism, it is tempting to speculate a synergic detrimental
neuropathic effect leading to the severe toxicity observed in our
patient. Of interest, we found electrophysiological findings consistent
with acute axonal dysfunction without pattern of demyelination. This
type of damage is the classic electrophysiological and
histopathological finding in beriberi neuropathy whereas arsenic axonal
toxicity characteristically coexists with segmental
demyelination.[14,15] An acute pattern of axonal damage has been
reported as a manifestation of arsenic poisoning but it has not been
observed as a side effect of short term administration of arsenic
trioxide at therapeutic dosage.[16] Thus, arsenic trioxide therapy may
lead to axonal loss through the exacerbation of the metabolic damage to
the nerve tissue induced by thiamine deficiency. This may have
relevance in considering prophylactic administration of thiamine to
subjects undergoing arsenic trioxide therapy for APL, a clinical
approach already adopted by others[7] and supported by animal models of
antioxidant properties of thiamine in arsenic treated animals.[17]
In
conclusion, occult thiamine deficiency may contribute to arsenic
toxicity and the combination may cause irreversible axonal neuropathy
which must be considered when monitoring APL patients on arsenic
trioxide therapy. Prophylactic administration of thiamine may be
considered in this setting.
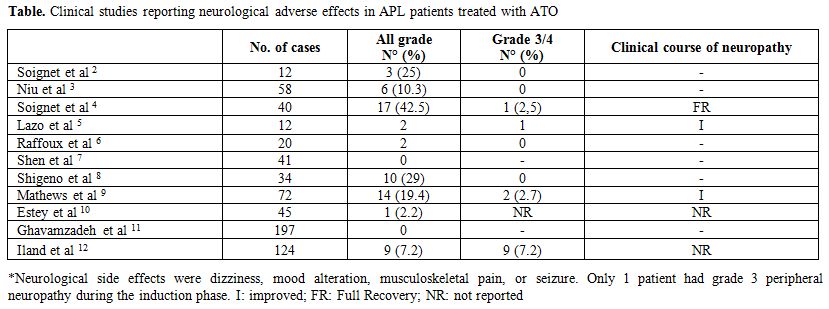 |
Figure 1. Clinical studies reporting neurological adverse effects in APL patients treated with ATO |
References

[TOP]