Received: February 3, 2016
Accepted: May 15, 2016
Mediterr J Hematol Infect Dis 2016, 8(1): e2016027, DOI 10.4084/MJHID.2016.027
This article is available on PDF format at:

Federica Sorà, Patrizia Chiusolo, Luca Laurenti, Francesco Autore, Sabrina Giammarco and Simona Sica
Istituto di Ematologia, Università Cattolica del Sacro Cuore di Roma, Roma.
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Life-threatening bleeding is a major
and early complication of acute promyelocytic leukemia (APL), but in
the last years there is a growing evidence of thromboses in APL. We
report the first case of a young woman with dyspnea as the first
symptom of APL due to massive pulmonary embolism (PE) successfully
treated with thrombolysis for PE and heparin. APL has been processed
with a combination of all-trans retinoic acid (ATRA) and arsenic
trioxide (ATO) obtaining complete remission. |
Case Report
Bleeding is the usual manifestation of acute promyelocytic leukemia
(APL) related coagulopathy and is associated with significant morbidity
and mortality.[1-3] Thrombocytopenia,
hyperfibrinolysis and disseminated intravascular coagulation (DIC) are
the leading causes of bleeding at diagnosis and shortly after the
introduction of treatment.[4] On the contrary
thrombotic complications were considered rare before the introduction
of ATRA. As a result, while the APL-related bleeding diathesis has been
investigated extensively and the pathogenesis has been elucidated to a
greater extent, the importance of thrombosis in APL, as a cause of
morbidity and mortality, has been undervalued and scarcely studied. So,
the exact pathogenetic mechanism of thrombosis in APL is still a matter
of controversy, even if the incidence of thrombotic complications in
APL seems to be rising. This could simply be due to higher levels of
vigilance and availability of advanced diagnostic modalities, or, due
to the increasing use of ATRA as suggested by Rashidi et al in a very
exhaustive review of this
topic.[5-7] There are
several potential explanations for thromboembolic complications in APL,
including: (i) A mere coincidence (e.g. genetic predisposition,
prolonged bed rest or immobility). (ii) Thrombogenicity of APL cells.
(iii) The use of all-trans retinoic acid (ATRA). (iv) The combination
of ATRA and antifibrinolytic agents, and the differentiation syndrome
caused by ATRA (i.e. ATRA syndrome).[4] We here
present the case of a young patient admitted to our hospital with a
massive pulmonary embolism as the first sign of acute promyelocytic
leukemia requiring thrombolysis and heparin. Finally, this case offers
a snapshot of the kinetic of APL related coagulopathy on therapeutic
heparin during ATRA and ATO and suggests the role of ATO and ATRA in
the treatment of APL patients with major thrombosis at diagnosis.
Methods and Result
A 24-year-old Caucasian female was admitted to the emergency
department of a peripheral hospital because of a few days history of
progressive shortness of breath. She also experienced an increasing
asthenia, uncommon for a young girl. Physical examination showed pallor
in the absence of bleeding, lymphadenopathy or hepatosplenomegaly. She
was afebrile and had only mild fluid retention. Vital signs including
blood pressure and heart rate were normal. Respiratory rate was 20
breaths per minute and O2 saturation by pulse oximetry was 96% with a FiO2
0.35 support by face mask. Her blood test revealed normocytic
normochromic anemia (Hb 7.7 g/dl) severe leucopenia and neutropenia
(WBC 800/mm3, ANC 480) with a platelet count of 109,000/mm3.
Peripheral smear did not show abnormal cells. Biochemistry values and
coagulation profile were normal. Chest X-Ray revealed ground-glass
opacity in right upper lobe.
The patient was referred 48 hours
later to our Division of Hematology. Sh complained of shortness of
breath requiring oxygen supplementation and she had two episodes of
hemoptysis. Her past medical history was unremarkable except for
adrenogenital syndrome requiring long-standing estroprogestinic support
and coeliac disease. She denied medication allergies, use of alcohol,
cigarettes or illicit substances. Neutropenia and anemia with a
platelet count of 110,000/mm3 were
confirmed and coagulation profile was normal except for d-dimer 7283
ng/ml (normal values <300ng/ml). International Society on Thrombosis
and Hemostasis (ISTH) DIC score was 4.[8]
Bone
marrow aspiration was carried out showing a massive infiltration of
pathologic promyelocytes and faggot cells consistent with APL. These
cells were markedly positive to myeloperoxidase. Flow cytometry
demonstrated an extensive positivity for CD117, myeloperoxidase (MPO),
CD45, CD13, CD33, and partial for CD34 and absence of HLA-DR, CD2
expression. CD15 was not tested. Fluorescence in situ hybridization
(FISH) revealed that 94% of nuclei were positive for t(15;17).
Cytogenetics showed a normal karyotype 46XX (20 cells). PML/RAR alpha
chimeric protein, bcr3 type, was detected by reverse
transcriptase-polymerase chain reaction (RT-PCR) from leukemic blasts.
FLT3 was positive.
Computerized tomography of the chest revealed a
massive acute pulmonary embolism with large filling defects of both
main arteries also involving lobar, segmental and subsegmental
arteries. There were also peripheral wedge-shaped areas of
hyperattenuation representing infarcts in many different lobes and a
minimal pericardial effusion was detected. The patient was started
immediately on intravenous sodium heparin and ATRA (45mg/m2 per day in two divided doses orally) with prophylactic methylprednisolone (0.5mg/kg) to prevent the differentiation syndrome.[10]
However, few hours later her general condition deteriorated with
hypotension, tachypnea (respiratory rate 32 breaths per minute) and
hypoxia with an oxygen saturation of 80% requiring a progressive
increase of FiO2 up to 1. Arterial blood gasses showed pH 7.477, pCO2 of 23.3 mm Hg, and pO2 of 59 mm Hg. Alteplase was given at the dose of 100 mg over two hours according to the recent guidelines from AACP.[9]
Sodium heparin was titrated in order to obtain 1.5 times the baseline
control value of the activated partial thromboplastin time (aPTT).
Bedside echocardiogram demonstrated mild right ventricular (RV)
hypertrophy with normal global kinesis. Pulmonary artery pressure
(PAPS) was estimated to be 31 mmHg. Lower limbs venous color Doppler
ultrasonography did not reveal signs of deep or superficial thrombosis.
Inherited and acquired thrombophilia screening tests were negative.
After thrombolysis, her pulmonary function dramatically improved.
At the second day of ATRA, considering the diagnosis of low-risk APL[10] and the requirement of long-term anticoagulation, arsenic trioxide (ATO) at the standard dose of 0.15mg/Kg/die was added.[11]
At the third day, breath rate was normal and oxygen supplementation was
discontinued. On day +11 of ATO+ATRA she developed mild chest pain,
asthenia and a slight increase in troponin levels and a T wave
abnormality on the ECG; chest CT scan was unchanged raising the
suspicion of a differentiation syndrome. ATRA was discontinued for five
days and dexamethasone 10mg bid was given until the complete resolution
of the symptoms. Leukocytosis, defined as a white-blood-cell count
(WBC) of more than 10×109 per liter, developed at day+ 17 and was successfully managed with hydroxyurea.[11] The kinetic of coagulative parameters, including platelets and WBC count during induction with ATO and ATRA, is depicted in figure 1.
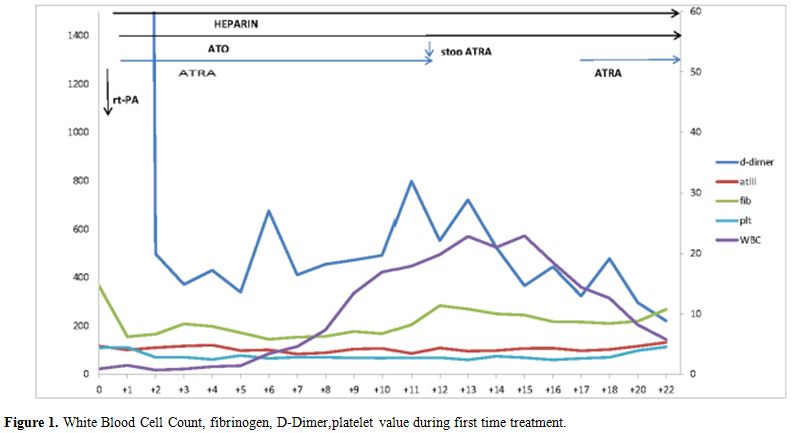 |
Figure 1. White Blood Cell Count, fibrinogen, D-Dimer,platelet value during first time treatment. |
After 50 days of ATRA and ATO peripheral blood cell count
and bone marrow biopsy documented the complete morphological and
molecular remission of APL and the patient was discharged from the
hospital on low molecular weight heparin. CT showed a slight reduction
of EP defects and the areas of hyperattenuation representing infarcts.
Echocardiogram described normal kinesis and valvular function with a
slight increased PAPS (35 mmHg). Consolidation therapy with ATRA and
ATO was delivered.[11]The patient is currently alive
and well after the end of ATO consolidation and in molecular CR at 20
months from diagnosis and off anticoagulation.
Discussion
Thrombotic events (TE) appear to be more common in acute
promyelocytic leukemia (APL) than in other acute leukemias, with
reported incidence ranging from 2 to 10-15%. Data from large
cooperative groups have been published after initial reports of
increased thrombotic events in APL when a combination of ATRA and
chemotherapy was applied.[12] The incidence of thrombosis in APL patients was 8.8% in AIDA protocol from the GIMEMA group[13-14] and 5.1% in PETHEMA LPA96 and LPA99 Protocol,[15] occurring mainly at diagnosis or during induction.[1]
In the GIMEMA study risk factors for the development of thrombosis
included higher leukocyte count, the prevalence of bcr3 transcript type
and a malignant cell phenotype including CD2 and CD15. ATRA-induced
differentiation syndrome was associated with a severe increase of
thrombotic complications in the PETHEMA group experience. These data
were partially confirmed in a recent paper from Mitrovic et al[16]
in a retrospective study of 63 APL patients. The authors reported an
incidence of thrombotic complications of 20.6%, including asymptomatic
thrombosis, and again more frequently observed during induction.
In
our case massive PE developed at diagnosis. No APL-specific risk
factors for thromboses or inherited or acquired thrombophilia were
found in this particular case except for bcr3 isoform and
estroprogestinic exposure. Despite the concurrent diagnosis of APL, the
patient required both heparin and thrombolysis for PE. Two other
attempts to treat major thromboses with thrombolysis were reported in
APL, one patient was treated for Budd-Chiari syndrome[17] and relapsed APL and a more recent one APL patient received local thrombolysis for arterial thrombosis at the onset of APL.[18] One additional patient underwent to thrombolysis for PE when in remission of APL.[19]
The combination of ATRA and ATO, adopted in considering the low-risk
APL category of our patient and the need for prolonged and sustained
anticoagulation, demonstrated to be an efficient regimen, also
significantly reducing the risk of prolonged thrombocytopenia due to
chemotherapy. Two additional cases with severe thrombotic events at
diagnosis successfully treated with anticoagulants with or without
thrombolysis when in ATO and ATRA have been published recently[18,20] and reported in Table 1.
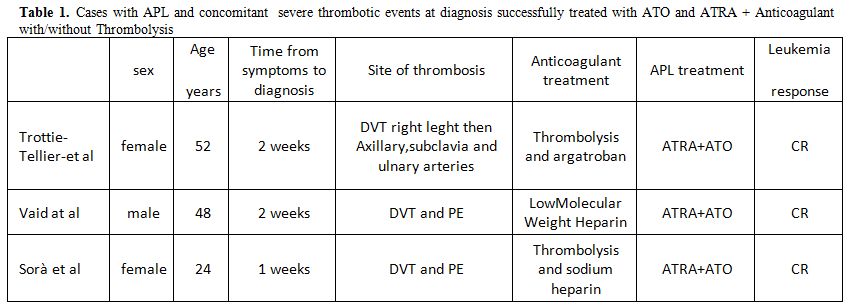 |
Table 1. Cases with APL and concomitant severe thrombotic events at diagnosis successfully treated with ATO and ATRA + Anticoagulant with/without Thrombolysis |
Although this case is exceptional, being to the best of our
knowledge the only reported case of thrombolysis for massive PE
attempted in APL at diagnosis, it raises several points of
discussion Thrombosis should be kept in mind for observation and
treatment of APL patients as suggested by a growing body of evidence.[1]
An accurate evaluation of risks should be pursued and anticoagulation
including thrombolysis might be necessary for APL at least in
catastrophic thrombosis. Although the management with ATRA and ATO in
induction, is largely anecdotal in APL patients anticoagulated, with
thrombosis and concurrent risk of bleeding, avoiding chemotherapy might
allow adequate anticoagulation by reducing the length and the severity
of thrombocytopenia. In literature now the results of some trials[21,22]
support the not inferiority of ATRA/ATO-based regimen in “all-risk”
acute promyelocytic leukemia regarding efficacy and safety; these data
and our experience suggest this combination also in high-risk APL
patients submitted to anticoagulation.
References

[TOP]