Hosein Kamranzadeh Fumani,
Mohammad Zokaasadi, Amir Kasaeian, Kamran Alimoghaddam, Asadollah
Mousavi, Babak Bahar, Mohammad Vaezi and Ardeshir Ghavamzadeh
Hematology, Oncology and Stem Cell Transplantation Research Center; Tehran University of Medical Sciences, Tehran, Iran.
Corresponding
author: Hosein Kamranzadeh Fumani, Hematology, Oncology and Stem Cell
Transplantation Research Center; Tehran University of Medical Sciences,
Tehran, Iran. Address: North karegar ave, Shariati hospital, Tel. &
Fax: +982188004140. E-mail:
Dr.kamranzadeh@gmail.com
Published: November 1, 2016
Received: August 3, 2016
Accepted: October 10, 2016
Mediterr J Hematol Infect Dis 2016, 8(1): e2016054, DOI
10.4084/MJHID.2016.054
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background and objectives:
Fanconi anemia (FA) is a rare genetic disorder caused by an impaired
DNA repair mechanism which leads to an increased tendency toward
malignancies and progressive bone marrow failure. The only curative
management available for hematologic abnormalities in FA patients is
hematopoietic stem cell transplantation (HSCT). This study aimed to
report the results of HSCT in adult or adolescent FA patients.
Patients and Methods:
Twenty FA patients with ages of 16 or more who underwent HSCT between
2002 and 2015 enrolled in this study. The stem cell source was
peripheral blood, and all patients had a full human leukocyte antigen
(HLA) matched donor, 19 patients had a sibling donor, and one had full
matched other related. Indications for HSCT were severe bone marrow
failure or dependence on blood products transfusion and failure of
medical treatment to sustain peripheral blood elements at an acceptable
level.
Results: Eleven
patients were female and 9 male (55% and 45%). Mean age was 24.05
years. Mortality rate was 50% (n=10), and the leading cause of death
was graft versus host disease (GVHD) which occurred in 5 patients (4
cases from acute GVHD and one from chronic GVHD). Survival analysis
showed an overall 5-year survival of 53.63% (95% confidence interval:
29.53%-72.74%) and 13 year survival of 45.96 % (95% confidence
interval: 22.08%-67.03%) among patients.
Conclusion:
HSCT is the only curative management for bone marrow failure in FA
patients. But the high rate of mortality and morbidity in adolescent
and adult patients makes it a challenging issue.
|
Introduction
Fanconi
anemia (FA) is a rare inherited disorder characterized by different
types of malformations, progressive bone marrow failure and an
increased tendency towards both hematological and solid malignancies.[1] FA arises from an underlying impaired DNA repair mechanism leading to chromosomal instability.[2]The
clinical manifestation of FA varies from case to case; therefore the
diagnosis may be delayed until the emergence of cytopenias.[3]
Diagnosis is confirmed with increased chromosomal breakage in cells
after exposure to DNA cross-linking agents such as diepoxybutane (DEB)
or mitomycin C.[1]There
are some methods available to control the disease as the use of
androgens, corticosteroids and supportive care such as transfusion of
blood products. But the only curative treatment offered for
hematological manifestations of FA is hematopoietic stem cell
transplantation (HSCT).[4-6] There are studies
available on the outcome of HSCT in FA patients, but most of them are
carried out on children. One of the important prognostic factors
related to the outcome of HSCT in FA patients is the recipients’ age.
It has been shown in some previous studies that advanced age is
associated with poorer outcome, more complications and consequently
higher mortality.[6,7] One of the most common complications of HSCT is acute GVHD which has a higher incidence in FA patients.[8] In this study, we focus on outcome and effects of HSCT on adolescents and adults with FA.
Patients and Methods
All
patients with a diagnosis of FA who were 16 years or older and
underwent HSCT between 2002 and 2015 were enrolled in this study. The
diagnoses were confirmed using mitomycin C sensitivity test which
showed increased chromosomal breakage and bone marrow examination was
performed including cytogenetic studies. All donors were screened for
FA. Covariates are extracted from the patients’ medical records and
included age, sex and other basic characteristics, malformations,
previous treatments, complete blood counts (CBCs) and bone marrow
cellularity, the result of cytogenetic studies, conditioning regimen
and acute and chronic GVHD.
Indications
for HSCT were severe bone marrow failure or blood product transfusion
dependency. Severe bone marrow failure defined as bone marrow
cellularity of less than 25% or cellularity of less than 50% with one
of the following; absolute neutrophil count (ANC) of less than
500/microliter, platelet count of less than 20000/microliter or
reticulocyte count of less than 60000/microliter. All transplants were
from a full HLA-matched donor; either sibling or other related, and the
source of stem cell was peripheral blood, and none were T-cell
depleted. Peripheral blood stem cell (PBSC) was chosen over bone marrow
because of feasibility, convenience, and acceptance by donors.
Neutrophil engraftment defined as ANC of greater than 500/microliter
for 3 consecutive days. No sex limits were set in this study. All
diagnostic tests (confirmation of FA), transplants and post-transplant
follow up visits took place in hematology and oncology and stem cell
transplant research center, Tehran University of medical sciences.
Informed consent was obtained from all patients at the beginning of the
study.
Continuous
variables were presented as mean values, and standard deviations and
categorical variables were shown as frequencies and percentages.
Overall survival was estimated by the Kaplan-Meier method and
accompanied with relevant 95% confidence intervals (CI). Median
follow-up time was computed with the reverse Kaplan-Meier method.
Univariate analysis of OS to calculate the hazard ratios (HR) of each
potential prognostic factors was performed using a Cox proportional
hazard regression. A p-value less than 0.05 was considered significant.
Data were analyzed using Stata software version 11.2
Results
Twenty
patients participated in our study. Mean age at transplant was 24.05
(SD=8.95) years ranging from 16 to 48 years. 45.0% patients were male
(n=9) and 55.0% were female (n=11). The frequency of malformations was
40% (8 cases) whom 6 cases (30.0%) had limb malformations, 1 had
nephron-urological malformation, and 1 had urogenital (5.0% each).Essential characteristics of studied population were briefed in table 1.
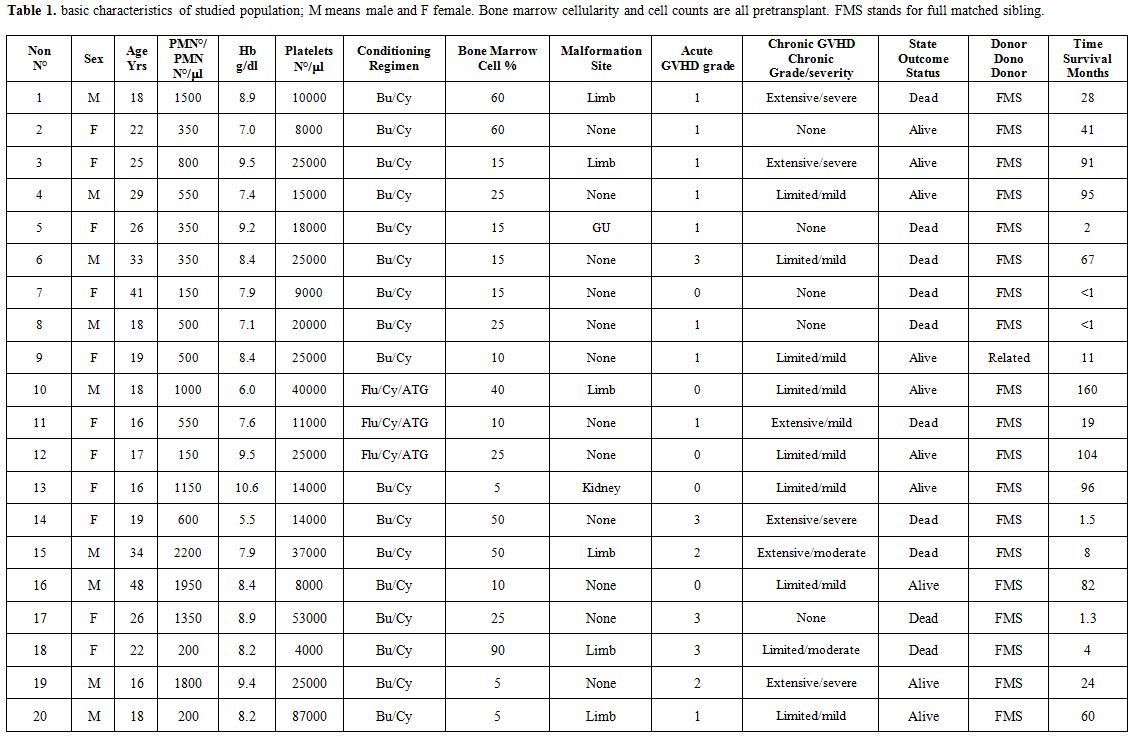 |
Table 1. Basic characteristics of studied
population; M means male and F female. Bone marrow cellularity and cell
counts are all pretransplant. FMS stands for full matched sibling. |
Fourteen
patients (70%) had severe bone marrow failure, and the remaining 6
(30%) were dependent on transfusion of blood products despite medical
treatments.Based
on clinical and pathologic findings there was no evidence of acute
leukemia or myelodysplastic syndrome. Cytogenetic studies in all
patients revealed no abnormalities.All
patients had a full HLA matched donor (19 siblings and one other
related). Seventeen patients received Bu/Cy; Busulfan: 0.2 mg/kg days 9
to day 6 and Cyclophosphamide 15mg/kg days 5 to day 2 before
transplantation. And 3 patients received Flu/Cy/ATG; fludarabine: 30mg/m2
for 5 days (days 9 to 5 before transplantation), cyclophosphamide 10
mg/kg for 2 days (day 4 and day 3 before transplantation) and equine
anti Thymocyte globulin (ATG) 10mg/kg; for days 4 to 1 prior to
transplantation as the conditioning regimen. GVHD prophylaxis for Bu/Cy
group consisted of methotrexate 10 mg/m2 for the first day after transplantation and 6 mg/m2
for days 3 and 6, cyclosporine 1.5 mg/kg intravenous days (IV) 3 to 7
after transplantation then 3 mg/kg IV from day 7 until the patient
tolerates oral, then changes to 5 mg/kg oral, and folinic acid 15 mg
oral days 2, 4, 5, 7, 8 and 9 after transplantation. For the Flu/Cy/ATG
group, GVHD prophylaxis was the only cyclosporine as mentioned.Median
follow up time was 26 months (maximum; 159 months).Graft failure
occurred only in one patient (5%) and median time to neutrophil
engraftment was 11 days after transplant.Fifteen
patients had acute GVHD, 9 patients were grade 1 and six were grades 2
to 4. There were 15 cases of chronic GVHD (9 limited and 6 extensive).Mortality
rate was 50% (10 out of 20 cases). Survival analysis using Kaplan-Meier
estimate were performed and showed a 5-year overall survival of 53.63%
(95% confidence interval: 29.53%-72.74%) Figure 1.
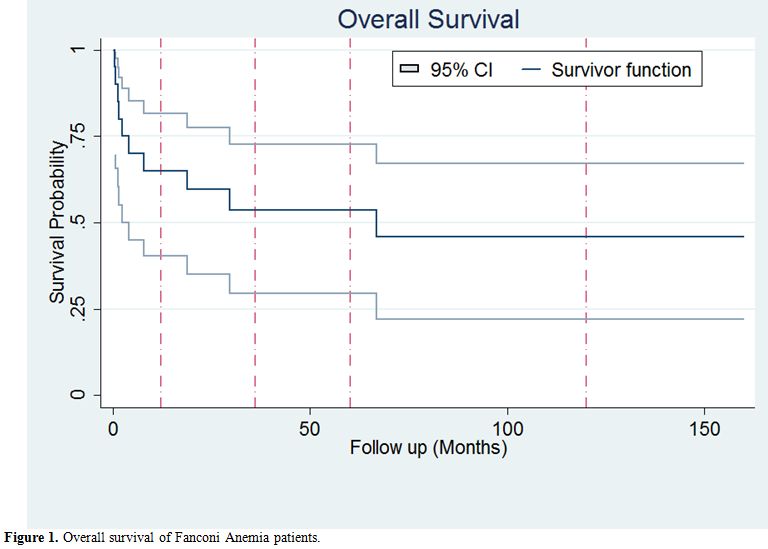 |
Figure 1. Overall survival of Fanconi Anemia patients. |
The leading cause of death was GVHD with 5 cases; 4 acute and 1 chronic, responsible for 50% of deaths reported.The
most frequent reason for death in the first month was sepsis (1 out of
2 cases died of sepsis, and the other died of sepsis and Diabetic
Ketoacidosis; DKA simultaneously) and for the 3-month was acute GVHD.
Other causes of death were hepatic mass (a mass lesion with radiologic
findings in favor of malignancy but the patient died before the biopsy
was taken) and renal failure.There is a significant relationship between mortality and grade 3 or 4 acute GVHD (log-rank; p-value=0.02).There
were no association between overall survival and age, sex, liver
function test, Hb and white blood cell, platelet count and bone marrow
cellularity before transplant (cox-regression).
Discussion
This retrospective study was aimed to assess the outcome of HSCT in adolescents and adults with Fanconi anemia.Acquired
idiopathic aplastic anemia (AA) syndromes are treated with HSCT as well
as FA. The report of Passweg et al. on severe AA patients underwent
HSCT showed 5-year OS of 66% during 1988-1992.[9]
Also, the report of Sangiolo et al. from Fred Hutchinson have shown
10-year OS of 65% in severe AA patients over the age of 40.[10]
In general, the survival of FA patients is lower than other severe AA
conditions. In a multicenter EBMT study in Europe, which analyzed the
results of 795 FA patients underwent HSCT from 1972 to 2010, with a
median follow up time of 6 years, an overall survival of 49% at 20
years was observed.[7] However, there are some reports
of better OS in transplanted FA patients. Bitan et al who transplanted
7 patients reported a survival of 100% and no severe acute GVHD with a
follow up time of 19 to 101 months,[11] similarly Tan
et al in a group of 11 FA patients with aplastic anemia (n=10) or MDS
(n=1) showed a 2 year overall and event free survival of 100% and 82%
respectively.[12] These above mentioned
studies, performed on small sample size showing a better outcome with a
short-term follow-up, include people with a median age at transplant
<10 years, before clonal evolution to leukemia/ MDS and, with
a conditioning fludarabine based regimens, without radiation.[7,12] Our study is comparable to EBMT results.[7]Previous
cooperative studies performed in a larger number of patients have shown
that older age, malformations, and leukemic evolution are associated
with worse outcome. In particular Guardiola et al. in a polycentric
study on patients with median age at transplantation of 10.8 years
(4.0-37.4 years), transplanted from unrelated donors, demonstrated the
poor outcome of patients with malformation 5 and the research of
Ayas et al., conducted on 3 groups of FA patients with MDS, acute
leukemia, and cytogenetic abnormality and a median age of 15, 18 and 13
years old, respectively, demonstrated that younger patients and
recipients of HLA-matched related donor transplantations with
cytogenetic abnormalities only have the best survival.[6]These
data was fundamentally confirmed by the large EBMT study in which a
better outcome was observed for patients transplanted before the age of
10 years, before clonal evolution (i.e., myelodysplastic syndrome or
acute myeloid leukemia), from a matched family donor, after a
conditioning regimen without irradiation, the latter including
fludarabine.[7] Furthermore, this study stressed the
long term negative affect associated with chronic GVHD, more frequent
in patients transplanted with PBSC.The
average age in our study was 24.05 years, and we also showed that
advanced age is associated to worse outcome which is similar to
previous reviews. Acute GVHD was detected in 75% of
patients during the follow up time, but most of them (9/15) had
grade 1 GVHD. AA
secondary to Fanconi should be distinguished from primary AA anemia.
Studies report lower incidence in severe AA patients. In Sangiolo et
al. avowed that subjects older than 40 years having HCT from
HLA-identical siblings had 35% acute GVHD of grade 2 or higher and 26%
chronic GVHD at 2-year;[10] similarly Passweg
et al. signaled 19% incidence of acute GVHD grade 2 to 4, and 32% of
chronic GVHD, at 5-year respectively in 1988-1992.[9]
Studies on FA patients showed a higher incidence. Peffault de Latour
revealed a 32% incidence of grade 2 to 4 acute and 14% and 19% 1-year
and 5-year chronic GVHD. Subjects with FA patients seem to be
more susceptible to developing acute GVHD, regardless of younger
age, rather than non-FA acquired aplastic anemia syndromes in response
to HSCT; the Guardiola et al. studies, performed in this field, have
shown a relative risk (RR) of 2.00 for FA patients[8] also a more significant effect of advanced age on GVHD incidence.[8]
The other factor which still remains controversial is the effect of
stem cell source, PBSC or bone marrow. Some reports advocate the higher
incidence of acute GVHD while some others like a Cochrane review showed
only lower acute GVHD grade 3 and 4 with bone marrow, but when grades 2
to 4 are analyzed the difference was not significant.[13] However, the chronic GVHD seems to be increased.[7]Our
study revealed a significant association between acute GVHD of grades 3
or more and higher mortality. Moreover, other clinically relevant
associations, as age and malformations do not reach a significant level
from the statistical point of view due to a limited number of patients,
typical of data of a rare disease reported by a single-center.
Furthermore, in our center, we perform the for Fanconi Anemia
only radiation-free therapy conditioning regimens which are shown
to be linked to fewer complications such as secondary malignancies
without compromising the engraftment.[12,14-15] Hence earlier studies suggested without-radiation protocols for conditioning FA patients to be considered.[16]
The rationale for the use of reduced intensity regimens is based on the
extreme vulnerability of FA patients to DNA damage which explains
better outcomes with RIC regimens and a good immunosuppression, which
probably is attained with busulfan and radiation free regimens and with
fludarabin and ATG.[19]The
primary cause of mortality in our study was GVHD (4 acute and 1
chronic) and infection which is comparable to former studies.[17]
Guardiola et al. suggested that the effect of on mortality is not
confined to severe acute GVHD in the first post-transplant months and
it also probably by inducing chronic GVHD can be a risk factor for
development of secondary malignancies, particularly in patients
transplanted with peripheral blood.[7,18] Experience with a conditioning regimen with fludarabine seems more favorable in term of GVHD and survival.[11,12,19]
Three of our patients were treated with this regimen, but a
confrontation in our set of patients is not possible due to a low
number of cases.The
outcome of HSCT in FA patients, in comparison to other idiopathic
acquired aplastic anemia syndromes, is less satisfactory due the
complications, and a higher mortality and morbidity rate. These
complications are more common in olders. It should be considered that
HSCT is still the only curative option for hematologic failure in these
patients. More studies focusing on new conditioning regimens, better
prophylaxis and management of GVHD including the use of different stem
cell sources (bone marrow rather than PBSC ) or T-cell depleted
methods may be needed to reach better results.
References
- Alter BP. Fanconi anemia and the development of
leukemia. Best Pract Res Clin Haematol. 2014;27(3-4):214-21.
http://dx.doi.org/10.1016/j.beha.2014.10.002 PMid:25455269
PMCid:PMC4254647
- Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003;97(2):425-40. http://dx.doi.org/10.1002/cncr.11046 PMid:12518367

- Stepensky
P, Shapira MY, Balashov D, Trakhtman P, Skorobogatova E, Rheingold L,
Brooks R, Revel-Vilk S, Weintraub M, Stein J, Maschan A, Or R, Resnick
IB. Bone marrow transplantation for Fanconi anemia using
fludarabine-based conditioning. Biol Blood Marrow Transplant .
2011;17(9):1282-8. http://dx.doi.org/10.1016/j.bbmt.2011.01.001 PMid:21220033
- MacMillan
ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia
- when and how? Br J Haematol. 2010;149(1):14-21. http://dx.doi.org/10.1111/j.1365-2141.2010.08078.x PMid:20136826
- Guardiola
P, Pasquini R, Dokal I, Ortega JJ, van Weel-Sipman M, Marsh JC, Ball
SE, Locatelli F, Vermylen C, Skinner R, Ljungman P, Miniero R, Shaw PJ,
Souillet G, Michallet M, Bekassy AN, Krivan G, Di Bartolomeo P,
Heilmann C, Zanesco L, Cahn JY, Arcese W, Bacigalupo A, Gluckman E.
Outcome of 69 allogeneic stem cell transplantations for Fanconi anemia
using HLA-matched unrelated donors: a study on behalf of the European
Group for Blood and Marrow Transplantation. Blood. 2000;95(2):422-9.
PMid:10627445
- Ayas M, Saber W, Davies
SM, Harris RE, Hale GA, Socie G, LeRademacher J, Thakar M, Deeg HJ,
Al-Seraihy A, Battiwalla M, Camitta BM, Olsson R, Bajwa RS, Bonfim CM,
Pasquini R, Macmillan ML, George B, Copelan EA, Wirk B, Al Jefri A,
Fasth AL, Guinan EC, Horn BN, Lewis VA, Slavin S, Stepensky P, Bierings
M, Gale RP. Allogeneic hematopoietic cell transplantation for fanconi
anemia in patients with pretransplantation cytogenetic abnormalities,
myelodysplastic syndrome, or acute leukemia. J Clin Oncol.
2013;31(13):1669-76. http://dx.doi.org/10.1200/JCO.2012.45.9719 PMid:23547077 PMCid:PMC3635221
- Peffault
de Latour R, Porcher R, Dalle JH, Aljurf M, Korthof ET, Svahn J,
Willemze R, Barrenetxea C, Mialou V, Soulier J, Ayas M, Oneto R,
Bacigalupo A, Marsh JC, Peters C, Socie G, Dufour C, Allogeneic
hematopoietic stem cell transplantation in Fanconi anemia: the European
Group for Blood and Marrow Transplantation experience. Blood.
2013;122(26):4279-86. http://dx.doi.org/10.1182/blood-2013-01-479733 PMid:24144640
- Guardiola
P, Socie G, Li X, Ribaud P, Devergie A, Esperou H, Richard P, Traineau
R, Janin A, Gluckman, E. Acute graft-versus-host disease in patients
with Fanconi anemia or acquired aplastic anemia undergoing bone marrow
transplantation from HLA-identical sibling donors: risk factors and
influence on outcome. Blood. 2004;103(1):73-7. http://dx.doi.org/10.1182/blood-2003-06-2146 PMid:12946993
- Passweg
JR, Socié G, Hinterberger W, Bacigalupo A, Biggs JC, Camitta BM,
Champlin RE, Gale P, Gluckman E, Gordon-Smith EC, Hows JM, Klein JP,
Nugent ML, Pasquini R, Rowlings PA, Speck B, Tichelli A, Zhang MJ,
Horowitz MM, Bortin MM. Bone marrow transplantation for severe aplastic
anemia: has outcome improved? Blood. 1997; 90(2): 858-64.
PMid:9226187
- Sangiolo D, Storb R,
Deeg HJ, Flowers ME, Martin PJ, Sandmaier BM, Kiem HP, Nash RA, Doney
K, Leisenring WM, Georges GE. Outcome of allogeneic hematopoietic stem
cell transplantation from HLA identical siblings for severe aplastic
anemia in patients over 40 years of age. Biol blood marrow tansplant.
2010; 16(10): 1411-8 http://dx.doi.org/10.1016/j.bbmt.2010.04.005 PMid:20403449 PMCid:PMC2934906
- Bitan
M, Or R, Shapira MY, Aker M, Resnick IB, Ackerstein A, Samuel S, Elad
S, Slavin S. Fludarabine-based reduced intensity conditioning for stem
cell transplantation of Fanconi anemia patients from fully matched
related and unrelated donors. Biol Blood Marrow Transplant: journal of
the American Society for Blood and Marrow Transplantation.
2006;12(7):712-8. http://dx.doi.org/10.1016/j.bbmt.2006.03.002 PMid:16785060
- Tan
PL, Wagner JE, Auerbach AD, Defor TE, Slungaard A, Macmillan ML.
Successful engraftment without radiation after fludarabine-based
regimen in Fanconi anemia patients undergoing genotypically identical
donor hematopoietic cell transplantation. Pediatr Blood Cancer.
2006;46(5):630-6. http://dx.doi.org/10.1002/pbc.20538 PMid:16078221
- Holtick
U, Albrecht M, Chemnitz JM, Theurich S, Skoetz N, Scheid C, von
Bergwelt-Baildon M. Bone marrow versus peripheral blood allogeneic
haematopoietic stem cell transplantation for haematological
malignancies in adults. The Cochrane database of systematic reviews.
2014(4):CD010189. http://dx.doi.org/10.1002/14651858.cd010189.pub2
- de
la Fuente J, Reiss S, McCloy M, Vulliamy T, Roberts IA, Rahemtulla A,
Dokal I. Non-TBI stem cell transplantation protocol for Fanconi anaemia
using HLA-compatible sibling and unrelated donors. Bone Marrow
Transplant. 2003;32(7):653-6. http://dx.doi.org/10.1038/sj.bmt.1704219 PMid:13130311
- Ayas
M, Siddiqui K, Al-Jefri A, El-Solh H, Al-Ahmari A, Khairy A, Markiz S,
Shahin H, Al-Musa A, Al-Seraihy A. Factors affecting the outcome of
related allogeneic hematopoietic cell transplantation in patients with
Fanconi Anemia. Biol Blood Marrow Transplant: journal of the American
Society for Blood and Marrow Transplantation. 2014;20(10):1599-603. http://dx.doi.org/10.1016/j.bbmt.2014.06.016 PMid:24960628
- Baker
JM, Lewis VA, Fernandez CV, Duval M, Crooks BN, Yuille K, Freedman MH,
Doyle JJ, Dror Y. Allogeneic hematopoietic stem cell transplantation of
patients with FA and high risk features using fludarabine without
radiation. Pediatr Blood Cancer. 2009;52(5):683-5. http://dx.doi.org/10.1002/pbc.21921 PMid:19156855
- de
Medeiros CR, Bitencourt MA, Zanis-Neto J, Maluf EC, Carvalho DS, Bonfim
CS, Funke VM, Setubal DC, Farah N, Pasquini R. Allogeneic hematopoietic
stem cell transplantation from an alternative stem cell source in
Fanconi anemia patients: analysis of 47 patients from a single
institution. Braz J Med Biol Res = Revista brasileira de pesquisas
medicas e biologicas / Sociedade Brasileira de Biofisica [et al].
2006;39(10):1297-304.
- Masserot C,
Peffault de Latour R, Rocha V, Leblanc T, Rigolet A, Pascal F, Janin A,
Soulier J, Gluckman E, Socie G. Head and neck squamous cell carcinoma
in 13 patients with Fanconi anemia after hematopoietic stem cell
transplantation. Cancer. 2008;113(12):3315-22. http://dx.doi.org/10.1002/cncr.23954 PMid:18831513
- Smetsers
SE, Smiers FJ, Bresters D, Sonnevelt MC, Bierings MB. Four decades of
stem cell transplantation for Fanconi anaemia in the Netherlands. Br J
Haematol. 2016 ;174(6):952-61. http://dx.doi.org/10.1111/bjh.14165
[TOP]