Salam Alkindi1,2, Saba AlMahrooqi2, Sumaiya AlHinai2, Ali AlMarhoobi1,2, Saif Al-Hosni2, Shahina Daar1,2, Naglaa Fawaz2 and Anil Pathare2
1 College of Medicine & Health Sciences, Sultan Qaboos University, Muscat, Oman
2 Department of Haematology, Sultan Qaboos University Hospital, Muscat, Oman
Corresponding
author: Dr. Salam Alkindi, BA, MB, BCh, BAO, DME, MSc, FRCP. Consultant
Haematologist, Department of Haematology, Professor, College of
Medicine & Health Sciences, Sultan Qaboos University, P. O.
Box 35, Muscat 123, Sultanate of Oman. Tel: +96-824411182, Fax:
+96-824413419. E-mail:
sskindi@yahoo.com
Published: February 15, 2017
Received: October 9, 2016
Accepted: January 9, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017013 DOI
10.4084/MJHID.2017.013
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Blood transfusion is an integral part of the supportive care for
patients with sickle cell disease (SCD) and thalassaemia. The hazard of
red cell alloimmunization, however, is one of the main complications of
this therapy.
Objectives:
The aim of this study was to evaluate the prevalence of red cell
alloimmunization in Omani patients with sickle cell anaemia and
thalassemia.
Methods: This
study included 262 patients whose historical transfusion records were
available. One hundred and twenty-nine patients with thalassaemia who
were attending the day care unit for regular transfusions, and 133 SCD
patients admitted at our hospital were included in this study. The
Diamed® gel system was used for the screening and identification of
atypical antibodies.
Results:
The rate of alloimmunization in SCD patients was 31.6% (n=42, 95%CI,
24.87-40.66), whereas in patients with thalassaemia it was 20% (n=26;
95%CI, 13.9-27.6). Antibodies to E, e, C, c, D, K, S, Fyª, Kpª, Jkª and
Cw were observed; 85% of the patients were also immunised with Rh and
Kell antigens. Considering the two groups together, 8 developed
nonspecific antibodies and 12 developed more than one antibody.
Conclusions:
Red cell transfusions were associated with a significant risk of
alloimmunization. It is, therefore, imperative to perform an initial
extended red cell phenotyping for both donors and recipients, and
carefully select ABO, Rh and Kell matched donors. The higher incidence
of alloimmunization in SCD patients is related to the inherent
SCD-specific inflammatory state.
|
Introduction
Sickle
cell disease and thalassaemia are the most frequent genetic disorders
in Oman with a combined carrier frequency rate of about 6%.[1-3]
Furthermore, in these congenital haemolytic disorders, there are
limited curative options. Thus, long-term blood transfusion remains an
integral treatment option for these conditions, in order not only to
save life but more importantly to improve the quality of life.[4]
Development of anti-RBC antibodies (alloantibodies and autoantibodies) can significantly complicate transfusion therapy.[5-7]
Furthermore, some of these alloantibodies being haemolytic, can cause
haemolytic transfusion reactions, and thereby limit the utility of
further transfusion, whereas others are clinically insignificant.[8] Erythrocyte
autoantibodies appear less frequently, but they can result in clinical
hemolysis and difficulty in cross-matching compatible blood units.[9]
Patients with autoantibodies may have a higher transfusion rate and
often require immunosuppressive drugs, splenectomy or alternative
treatments to maintain an adequate level of haemoglobin.
Despite the recognition of antibodies as a transfusion-associated risk,[7,10-13]
little is known about the extent and causes of these phenomena among
thalassaemia and sickle cell disease patients from the Sultanate of
Oman or the most appropriate methods of preGenetic blood disorders survey in the Sultanate of Oman
vention. Approaches for
prevention or treatment of alloimmunizations are under debate and
include the provision of RBCs matched for all the major antigens
associated with clinically significant antibodies, or to only give
blood matched for antibodies that have already been detected. The
reason for such a controversy may lie in the fact that many
alloantibodies are not harmful and that expensive prevention methods
may, therefore, benefit only some patients.[14] In
addition, donor feasibility and the cost of RBC matching could impact
on these approaches as also the own local guidelines regarding this
issue. Furthermore, a better knowledge basis of the potential harmful
antibodies among the thalassaemia and sickle cell disease patients can
assist in considering the appropriate transfusion strategy to use. Our
objective was to assess the prevalence of alloimmunization among our
multiply transfused patients with thalassaemia and sickle cell anaemia.
Materials and Methods
Diagnosis
of homozygous thalassaemia major and sickle cell disease was initially
made by high-performance liquid chromatography [HPLC] profiles.
However, it was further confirmed with family member studies [parents]
and where necessary, by DNA studies using Sanger sequencing.
Thalassemia patients:
Clinical features and transfusion records of 129 thalassaemia patients,
aged 5-32 years, 44 males, 85 females, who received regular transfusion
were analysed. These patients were attending the day care unit at SQUH
for regular transfusions.
Sickle cell anaemia patients:
133 sickle cell disease patients [113 SS and 20 S-beta thal] who were
admitted to SQUH haematology wards (30 males and 103 females) and who
received regular transfusion were analysed. The transfusion records of
all the patients including those transfused for their first time were
examined for the presence of alloimmunization and antibody specificity,
age, gender and ethnicity.
Donors:
Blood donors from the SQUH blood bank were identified for their racial
background, and RBC phenotype was performed for the following antigens
C, c, D, E, e and Kell. The donor’s ethnic origin was classified
into Arabs and non-Arabs.[Data not shown]
Laboratory protocol. Antibody screening:
Detection of alloantibodies was performed on a fresh blood sample using
the indirect antiglobulin test by the column agglutination method. The
gel card centrifugation technique was used (DiaMed AG, Cressier sur
Morat, Switzerland). All patients were screened before any transfusion.
Antibody identification:
Antibody specificity was determined using a standard panel of red cells
reacting to known antigens using column agglutination and gel
centrifugation (ID-DiaPanel and ID-DiaPanel-P, DiaMed AG). The indirect
antiglobulin test and enzymatic papain–treated RBC test at 370C were
performed when necessary and elution of antibodies was done to help in
identification. Detection of alloantibodies masked by autoantibodies
required the use of adsorption techniques using Polyethylene glycol
[PG], or albumin or low-ionic strength saline [LISS] to identify the
underlying antibody by the indirect antibody test [IAT].
Results
Thalassaemia:
26(20%) of the 129 patients had positive antibody screening, in whom 34
IgG alloantibodies were detected (95%CI, 13.9-27.6). 18(69%) patients
developed one antibody; 6(23%) developed two antibodies and one (4%)
developed three antibodies. One of the patients presented with a
non-specific antibody (NSA). Table 1 and Figure 1b
shows the specificities and frequencies of the alloantibodies; 30(88%)
of the alloantibodies were against the Rh and Kell antigens. The rate
of alloimmunization among males was 19% and females 21%.
The rate of alloimmunization in adults (aged 13-32 years) was higher at 14.4% as compared to 4% in children (aged 5-12 years).
Sickle cell anaemia:
Out of 133 patients, 42(31.5%) developed positive antibody screen in
whom 46 IgG alloantibodies were detected (95%CI, 24.87-40.66). Seven
patients (16.6%) showed NSA, 23(54.7%) developed one antibody; 10(24%)
developed two antibodies, and two patients had developed three
antibodies. The specificities and frequencies of the alloantibodies in
Omani patients with SCD are shown in Table 1 and Figure 1a.
38(83%) of the alloantibodies were Rh and Kell antibodies. The rate of
alloimmunization among males was 30% and among females 33%.
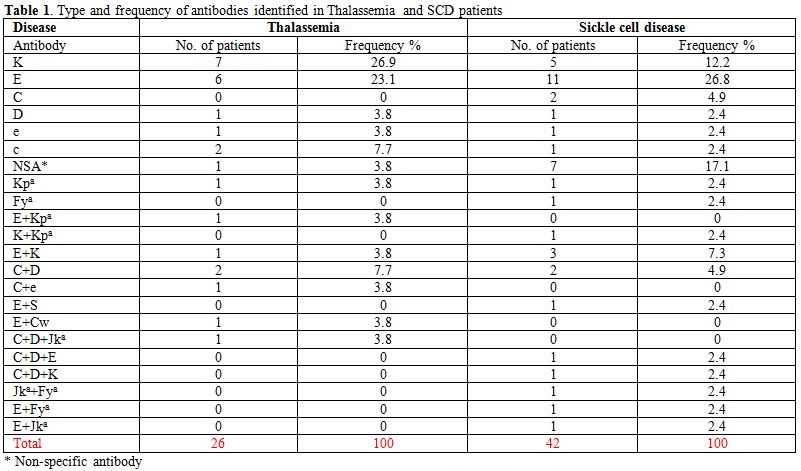 |
Table1. Type and frequency of antibodies identified in Thalassemia and SCD patients |
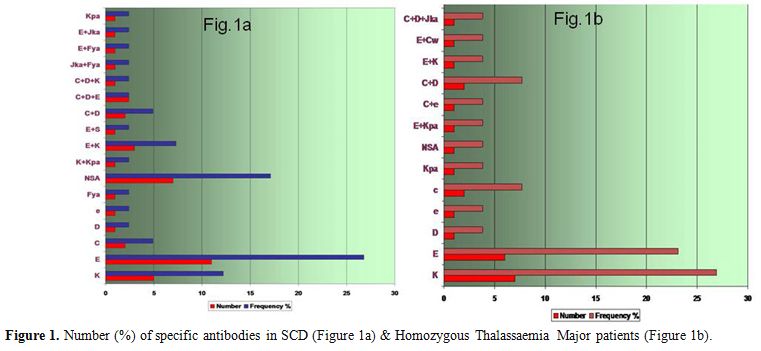 |
Figure 1. Number (%) of specific antibodies in SCD (Figure 1a) & Homozygous Thalassaemia Major patients (Figure 1b). |
Furthermore,
in the eight patients who had a non-specific antibody, we observed that
PG-IAT detected clinically significant antibodies like anti-E, anti-C,
anti-D, anti-Jk(a), anti-c, anti-e, anti-s that were masked by an
autoantibody in our cohort of multi-transfused patients. PG-IAT was
superior in detecting clinically significant allo- antibodies in the
presence of masking autoantibodies as compared to the other techniques
employed.
Discussion
The
factors involved in alloimmunization are complex and includes at least
three main contributing elements: the RBC antigenic differences between
the blood donor and recipients, the recipient’s immune status and the
immunomodulatory effect of the allogeneic blood transfusions on the
recipient’s immune system.[15,16]
This study
shows that the prevalence of alloimmunization in Omani homozygous
thalassaemia patients was 20% (n=26; 95%CI, 13.9-27.6). Comparing the
rate of alloimmunization in Omani thalassaemia patients with that of
other populations, it was similar to several countries namely, 16.32%
from Iran,[17] and 22% from California[18] and 19% from the CDC data in the USA on Asian and Caucasians patients.[19]
But in general, there is a reduction of the frequency of
alloimmunization when the patient receives blood from the same ethnic
groups like those living in Hong Kong[20] and in Saudi Arabia.[21]
The low incidence of immunization found in an old Italian cooperative
study of 1984 (5/68;5.4%), could have the same explanation since this
study included only thalassaemic patients living in Italy and receiving
blood from the same ethnic group.[22] The higher rate
of alloimmunization in adults as compared to children is in keeping
with other studies where age is a significant factor.[11]
The incidence of alloimmunization in transfused Omani patients with SCD
was found to be 31.5% (n=42, 95%CI, 24.87-40.66). This rate is similar
to that reported in USA, France, Holland, and patients of Asian descent
in Brazil.[23-27] It was also noticed that most of the alloantibodies were to Rh and Kell, antigens and that the E antibody had a higher rate.
The
Omani population, for historical reasons, is known to be a mixture of
more than one ethnic group. So it was expected to detect some antigenic
differences among the Omani donors themselves. Also, 10% of donors are
non-Omani, so patients receiving blood transfusions will be further
exposed to “foreign” antigens. The frequency of alloantibodies may be
reduced by limiting the transfusion from donors with the same ethnic
origin.[21]
It was noticed that patients with
sickle cell anaemia showed a slightly higher rate of alloimmunization
(31.5%) than thalassaemia patients (20%). This datum is consistent with
observations by other studies as well. One reason for this observation
could be because thalassemia patients are usually transfused at a
younger age and regular intervals. The immune system response will be
affected by the patient’s age at their first transfusion and number of
blood units the patient received.[27] It is believed
that transfusions at an early age may offer some immune tolerance and
protection against alloimmunization. The relation between the number of
blood units transfused and antibody formation is unknown in
thalassemia, but it is a major factor for increased alloimmunization in
patients, including SCD, who receive multiple transfusions. However it
should be taken into account that SCD is a chronic inflammatory state,
and pro-inflammatory stimuli promote alloimmunization.[28,29]
Furthermore, age is a significant factor, so children with SCD, who are
chronically transfused, might have less inflammation, which could
explain their lower rate of alloimmunization.[30,31]
However, some antigen-negative patients may not produce antibodies at
all or may form only one antibody despite exposure to antigen-positive
cells. Studies have suggested at least in SCD patients, that genetic
makeup is very relevant to the development of antibodies mainly altered
Rh or Kell alleles, and perhaps acquiring these antibodies may be
genetically driven.[32,33] In Omani population
further studies will be needed to assess the effect of the number of
transfusions on the immune response, the effect of the age at which the
patient is first transfused, and the genetic makeup of recipients and
donors on alloimmunization.
One of the biggest problems in a
conventional hospital blood bank is finding the appropriate
antigen-negative blood for the allo-immunised patients. Numerous
reports show that transfusion of phenotype-matched RBCs (Rh and Kell)
can reduce the risk.[19,34]
However, there are still few reports revealing that the risk of
alloimmunization is still high even when the donor blood is Rh and Kell
matched with the recipient.[12,13]
BCSH
transfusion guidelines also state that all patients with sickle cell
disease and thalassaemia have their full phenotype tested at diagnosis
and are given matched blood for C, c, E, e and K.[35,36]
Moreover, extended red cell phenotype matching, although useful in
preventing the formation of most alloantibodies, may prove impractical
to provide adequate and timely donors for these patients.[37]
At present, we too follow this standard recommendation and hope
to decrease the rate of alloimmunization. In our hospital it is
estimated that the cost of one year of phenotyping for Rh and Kell
antigens is about 12,500 OMR ($32,400) for all the donated units in our
blood bank, raising the question whether it is cost effective to
phenotype all of these units routinely. Nevertheless, DNA-based
phenotyping can overcome certain limitations of serological studies and
is beneficial in patients recently transfused or with interfering allo-
or autoantibodies.[38]
Conclusions
Red
cell transfusions are the cornerstone in the management of homozygous
thalassaemia major but remain underutilised in SCD patients for fear of
complications, although they can be lifesaving in the context of SCD
complications. Nonetheless, they are associated with a considerable
risk of alloimmunization as well as iron overload. The current BCSH
transfusion guidelines recommend the initial extended red cell
phenotyping for both donors and recipients. Thus with careful selection
of donated units, coupled with further elaborative studies of the
genetic diversity of patients and donor pool will certainly go a long
way in reducing the prevalence of red cell alloimmunization. Therefore
it will be cost-effective in the long term to choose the appropriate
blood donor
Acknowledgement
We wish to thank the hospital administration for the use of hospital material for this study.
References
- Al-Riyami A, Ebrahim GJ, Genetic
blood disorders survey in the Sultanate of Oman. J Trop. Pediatr.
2003 Jul; 49 suppl 1: i1-20

- Al-Riyami
AA, Sulieman AJ, Afifi M, Al-lamki ZM, Daar S, A community- based study
of common hereditary blood disorders in Oman. East Mediterr. Health J
2001 Nov; 7(6):1004-11
- Alkindi S, Al Zadjali S, Al Madhani A, Daar S,
Al Haddabi H, Al Abri Q, Gravell D, Berbar T, Pravin S, Pathare A,
Krishnamoorthy R. Forecasting hemoglobinopathy burden through neonatal
screening in Omani neonates. Hemoglobin. 2010; 34: 135-44. http://dx.doi.org/10.3109/03630261003677213
- Wayne AS, Kevy SV, Nathan DG, Transfusion management of sickle cell disease. Blood 1993; 81:1109-23
- Hmida S, Mojaat N, Maamar M, Bejaoui M,
Mediouni M, Boukef K, Red cell alloantibodies in patients with
haemoglobinopathies. Nouv Rev Fr Hematol 1994 oct; 36 (5): 363-6.
- Spanos T, Karageorga M, Ladis V, Peristeri J,
Hatziliami A, Kattamis C. Red cell alloantibodies in patients with
thalassaemia. Vox Sang 1990; 58( 1): 50-5. http://dx.doi.org/10.1111/j.1423-0410.1990.tb02055.x
- Rosse WF, Gallagher D, Kinney T, Castro O,
Dosik H, Moohr J,Wang W, Levy PS, Transfusions and alloimmunization in
sickle cell disease. The cooperative study of sickle cell disease.
Blood 1990 Oct.1; 76(7): 1431-7.
- Aygun B, Padmanabhan S, Paley C,
Chandrasekaran V. Clinical significance of RBC alloantibodies and
autoantibodies in sickle cell patients who received transfusions.
Transfusion 2002 Jan; 42(1): 37-43. http://dx.doi.org/10.1046/j.1537-2995.2002.00007.x
- Wayne AS, Kevy SV, Nathan DG, Transfusion management of sickle cell disease. Blood, 1993 Mar 1; 81(5): 1109-23.
- Castellino SM, Combs MR, Zimmerman SA, Issitt
PD, Ware RE, Erythrocytes autoantibodies in paediatric patients with
sickle cell disease receiving transfusions therapy: frequency,
characterization, and significance. Br. J. Haematol 1999 Jan;
104(1): 189-94. http://dx.doi.org/10.1046/j.1365-2141.1999.01127.x
- Murao M, Viana MB, Risk factors for
alloimmunization by patients with sickle cell disease. Braz J. Med.
Biol Res 2005; 38: 675-682 http://dx.doi.org/10.1590/s0100-879x2005000500004
- Chou ST, Jackson T, Vege S, Smith-Whitley K,
Friedman DF, Westhoff CM. High prevalence of red blood cell
alloimmunization in sickle cell disease despite transfusion from
Rh-matched minority donors. Blood. 2013;122(6):1062-71. https://doi.org/10.1182/blood-2013-03-490623
- Miller ST, Kim H-Y, Weiner DL, Wager CG,
Gallagher D, Styles LA, et al. Red blood cell alloimmunization in
sickle cell disease: prevalence in 2010. Transfusion. 2013;53(4):704-9.
https://doi.org/10.1111/j.1537-2995.2012.03796.x
- Wayne As, Schoenik SE, Pegelow CH, Financial
analysis of chronic transfusions for stroke prevention in sickle cell
disease. Blood 2000 Oct.1; 96(7):2369-72
.
- Alarif L, Castro O, Ofosu M, Dunston
G, Scott RB, HLA –B35 is associated with red cell alloimmunization in
sickle cell disease. Clin immunol Immunopathol 1986, 38:178-183. http://dx.doi.org/10.1016/0090-1229(86)90136-4
- Vichinsky EP, Earles A, Johnson RA,
Hoag MS, Williams A, Lubin B, Alloimmunization in sickle cell anemia
and transfusion of racially unmatched blood. N Eng J Med 1990, 322:
1617-21. http://dx.doi.org/10.1056/nejm199006073222301
- Davari K, Soltanpour MS. Study of
alloimmunization and autoimmunization in Iranian beta-thalassemia major
patients. Asian J Transfus Sci. 2016 Jan-Jun;10(1):88-92. http://dx.doi.org/10.4103/0973-6247.172179
- Singer ST, Wu V, Mignacca R, Kuypers FA,
Morel P, Vichinsky EP Alloimmunization and erythrocyte autoimmunization
in transfusion-dependent thalassemia patients of predominantly Asian
descent. Blood. 2000;96:3369-3373.CrossRefMedlineWeb of ScienceGoogle
Scholar
- Vichinsky E, Neumayr L, Trimble S, Giardina
PJ, Cohen AR, Coates T, Boudreaux J, Neufeld EJ, Kenney K, Grant A,
Thompson AA; CDC Thalassemia Investigators.Transfusion complications in
thalassemia patients: a report from the Centers for Disease Control and
Prevention (CME). Transfusion. 2014 Apr;54(4):972-81; quiz 971. http://dx.doi.org/10.1111/trf.12348
- Ho HK, Ha SY, Lam CK, Chan GC, Lee TL, Chiang
AK, Lau YL. Alloimmunization in Hong Kong southern Chinese
transfusion-dependent thalassemia patients. Blood.2001 Jun
15;97(12):3999-4000. PubMed PMID: 11405212
- Abdel Gader AM, Al Ghumlas AK, Al-Momen AM,
Transfusion medicine in a developing country-alloantibodies to red
blood cells in multi-transfused patients in Saudi Arabia. Transfus
Apher Sci.2008 ;39:199-204. http://dx.doi.org/10.1016/j.transci.2008.09.013
- Sirchia G, Zanella A, Parravicini A, Morelati
F, Rebulla P, Masera G, Red cell alloantibodies in thalassemia major:
results of an Italian cooperative study. Transfusion 1985 Mar-Apr;
25(2)110-2. http://dx.doi.org/10.1046/j.1537-2995.1985.25285169198.x
- Vichinsky EP, Current issues with blood transfusion in sickle cell disease. Semin. Hematol 2001 Jan ; 38( 1 suppl 1): 14-22 http://dx.doi.org/10.1053/shem.2001.20140
- Moreira Junior G, Bordin JO, Kuroda A,
Kerbauy J, Red cell alloimmunization in sickle cell disease, the
influence of racial and antigenic pattern differences between
donors and recipients in Brazil. Am. J Hematol 1996 Jul; 52(3) :
197-200. http://dx.doi.org/10.1002/(sici)1096-8652(199607)52:3<197::aid-ajh11>3.0.co;2-d
- Sins JW, Biemond BJ, van den Bersselaar SM,
Heijboer H, Rijneveld AW, Cnossen MH, Kerkhoffs JL, van Meurs AH, von
Ronnen FB, Zalpuri S, de Rijke YB, Ellen van der Schoot C, de Haas M,
van der Bom JG, Fijnvandraat K. Early occurrence of red blood cell
alloimmunization in patients with sickle cell disease. Am J Hematol.
2016 Aug;91(8):763-9. http://dx.doi.org/10.1002/ajh.24397
- Norol F, Nadiahi J, Bachir D, Desaint C,
Guillou Bataille M, Beajean F, Bierling P, Bonin P, Galacteros F,
Duedari N, Transfusion and alloimmunization in sickle cell anemia
patients. Transfus Clin Biol. 1994; (1): 27-34. http://dx.doi.org/10.1016/s1246-7820(05)80054-0
- Murao M, Viana MB, Risk factors for
alloimmunization by patients with sickle cell disease. Braz J Med Biol
Res 2005 May; 38(5) : 675-82. http://dx.doi.org/10.1590/s0100-879x2005000500004
- Yu J, Heck S, Yazdanbakhsh K. Prevention of
red cell alloimmunization by CD25 regulatory T cells in mouse models.
Am J Hematol. 2007;82(8):691–696. https://doi.org/10.1002/ajh.20959
- Hendrickson JE, Chadwick TE, Roback JD,
Hillyer CD, Zimring JC. Inflammation enhances consumption and
presentation of transfused RBC antigens by dendritic cells. Blood.
2007;110(7):2736–2743. https://doi.org/10.1182/blood-2007-03-083105
- Murao M, Viana MB. Risk factors for
alloimmunization by patients with sickle cell disease. Braz J Med Biol
Res. 2005;38(5):675–682. https://doi.org/10.1590/s0100-879x2005000500004
- Gill
FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, Pegelow CH,
Vichinsky E. Clinical events in the first decade in a cohort of infants
with sickle cell disease: Cooperative Study of Sickle Cell Disease.
Blood. 1995;86(2):776–783.
- Chou ST, Westhoff CM, Molecular biology of
the Rh system: clinical consideration for transfusions in sickle cell
disease. Am Soc Hematol Educ program 2009: 178-84. http://dx.doi.org/10.1182/asheducation-2009.1.178
- Boturao-Neto E, Chiba AK, Vicari P,
Figueiredo MS, Bordin JO, Molecular studies reveal a concordant KEL
genotype between patients with hemoglobinopathies and blood donors in
Sao Paulo City Brazil. Haematologica 2008 sep;93(9): 1408-10. http://dx.doi.org/10.3324/haematol.12766
- Vichinsky EP, Luban NL, Wright E, Olivieri N,
Driscoll C, Pegelow CH, Adams RJ, Prospective RBC Phenotype matching in
a stroke –prevention trial in sickle cell anemia :a multicenter
transfusion trail. Transfusion 2001 Sep; 41(9):
1086-92. http://dx.doi.org/10.1046/j.1537-2995.2001.41091086.x
- Guidelines for pre-transfusions compatibility
procedures in blood transfusions laboratories-BCSH Blood Transfusions
Task Force. Transfus Med. 1996 Sept; 6(3):273-83. http://dx.doi.org/10.1111/j.1365-3148.1996.tb00079.x
- Vichinsky EP, Ohene-Frempong K, Thein SL,
Lobo CL, Inati A, Thompson AA, Smith-Whitley K, Kwiatkowski JL,
Swerdlow PS, Porter JB, Marks PW, Transfusion and chelation
practices in sickle cell disease: a regional perspective. Pediatr
Hematol Oncol 2011 Mar; 28(2): 124-33. http://dx.doi.org/10.3109/08880018.2010.505506
- Castro O, Sandler G, Houston –Yu P, Rana S,
Predicting the effect of transfusing only phenotype –matched RBC to
patients with sickle cell disease: theoretical and practical
implications. Transfusions 2002 Jun; 42:684-90. http://dx.doi.org/10.1046/j.1537-2995.2002.00126.x
- Fasano RM, Chou ST. Red Blood Cell Antigen
Genotyping for Sickle Cell Disease, Thalassemia, and Other Transfusion
Complications. Transfus Med Rev. 2016 Oct;30(4):197-201. http://dx.doi.org/10.1016/j.tmrv.2016.05.011
[TOP]