M. Al Huneini1, S. Alkindi1,2, V. Panjwani1, K. Al Falahi1, B. Al Balushi1, D. Gravell1, C.H. Ho1, R. Krishnamoorthy3 and A.V. Pathare1
1 Sultan Qaboos University Hospital, Muscat, OMAN.
2 Sultan Qaboos University, College of Medicine and Health Sciences, Muscat, OMAN.
3 INSERM, U665, F-75015 Paris, France; Laboratoire d’Excellence GR-EX, Paris, France.
Corresponding
author: Dr. Anil Pathare, MD, FCPS, FIMSA, PhD. Senior Consultant
Haematologist, Department of Haematology, Sultan Qaboos University
Hospital, P. O. Box 35, Muscat 123, Sultanate of Oman. Tel:
+96824144906, Fax: +96824144887. E-mail:
avp16@hotmail.com
Published: April 20, 2017
Received: December 21, 2016
Accepted: March 23, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017028 DOI
10.4084/MJHID.2017.028
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Objectives: To
explore the incidence of vaso-occlusive crisis (VOC) in Blood Group “O”
sickle cell disease (SCD) patients, and correlate it with the blood
group and thrombospondin (TSP) levels.
Methods:
In 89 consecutive SCD patients, blood samples were obtained for von
Williebrand factor (vWF:Ag) antigen, collagen binding activity
(CBA), ristocetin binding activity (RCo), blood group typing,
C-reactive protein (CRP), high performance liquid chromatography
(HPLC), Serum TSP 1 and TSP 2 levels, complete blood counts (CBC),
lactic dehydrogenase (LDH) levels, liver function (LFT) and renal
function tests (RFT) during VOC episodes and in steady state
conditions.
Results: In
steady state SCD patients (n=72), “O” blood group patients (n=37)
showed a significantly higher median serum TSP 1 and TSP 2 levels as
compared to non-O blood group patients [n=35] [p <0.05, Mann-Whitney
test]; with an inverse relation between vWF:Ag, Factor VIII:C and TSP
levels. Furthermore, the serum TSP 1 and TSP 2 levels were
significantly higher in patients presenting with acute VOC [n=17], as
well as in those with repeated VOC’s (group 1, n=16), especially
amongst blood group “O” patients [p, <0.05, Mann-Whitney test].
Conclusions:
The study demonstrates an inverse relation between TSP and vWF levels,
in blood group “O” SCD patients, with an upregulation of the TSP
levels. Expectedly, during active VOC crisis, the TSP 1 and TSP 2
levels were significantly elevated.
|
Introduction
Sickle cell disease (SCD) is a condition with protean manifestations and demonstrates considerable clinical variability.[1,2] The disease constitutes one of the most frequent causes of hospitalizations in the Sultanate of Oman.[3,4]
It is characterized by chronic hemolysis, frequent infections,
recurrent occlusion of microcirculation; leading to painful crises,
chronic organ damage and premature death. Intermittent painful episodes
due to the vaso-occlusive crisis (VOC) is the most common clinical
manifestation of SCD, but subclinical episodes also occur.[5]
The mechanisms by which VOC’s are initiated is complex and multifactorial.[5-9]
Sickle red blood cells (RBCs) contribute to the initial VOC process and
play a significant part in nearly all the clinical manifestations of
SCD. The pro-adhesive sickle cells bind to endothelial cell P-selectin,
E-selectin, intercellular adhesion molecule-1, vascular cell adhesion
molecule-1, CD36, leading to the complex process of endothelial
activation.[8-11] Polymerization of deoxy-HbS is an
ongoing process in SCD and plays the most significant role in the
process of sickling of RBCs in SCD.[12] Thus, the end
result of multiple episodic cycles of polymerization of deoxy-HbS with
dehydration of the RBC is a dense, irreversibly sickled red cell.
However, when oxygenated, an irreversibly sickle cell may contain no
polymer but is nonetheless distorted in shape and may still contribute
to vaso-occlusion.[12-14] These features make negotiation of the microvasculature difficult, if not impossible, for these sickle erythrocytes.[14]
RBCs
in SCD also appear to have an increased binding affinity to the
vascular endothelium. The degree of affinity correlates strongly with
the severity of clinical disease. Several molecular interactions
contribute to this endothelial affinity and are mediated by increased
levels of integrin VLA-4 (α4β1)[15] and membrane glycoprotein IV (CD36).[16]
VLA-4 mediates adhesion both to endothelial vascular cell adhesion
molecule-1 (VCAM-1) and to fibronectin present in activated
endothelium, whereas, CD36 mediates adhesion via thrombospondin to αVβ3
integrin on activated endothelium. Thrombospondin is normally present
in platelet α granules and is released from activated platelets.[15,16] Thrombospondin binds CD47 (integrin-associated protein) expressed on RBC membranes in addition to binding with CD36.[7,17]
The endothelial selectin activation by adhesion molecules expressed in
sickle red cells, and their inhibition of endothelium-dependent
vasorelaxation, by blocking the endothelium-derived relaxing factor
(EDRF)[18,19,20] contribute to worsening
vaso-occlusion. Further, it has long been known that the
microvasculature of patients with SCD may develop intimal hyperplasia.
This creates irregular areas of endoluminal narrowing, which worsen
vaso-occlusion by promoting thrombosis. This process has been
documented in the cerebral and splenic vascular beds.[21]
Many factors are known to affect the frequency of VOC, such as HbF concentration, sickle βs
haplotypes, and the presence of various adhesive substances, which
enhance the sickle red cells adherence to the subendothelial
structures. TSP and von Willebrand factor (vWF) are among the proteins
that have been implicated as mediators of the adhesive interactions
between sickle erythrocytes and the blood vessel wall.[22-24] However, sickle erythrocytes were found more adherent to immobilized TSP than to vWF.[25]
Furthermore, TSP1 has been involved in the liberation of toxic membrane
vesicles from RBCs, which contributes to the degradation of vascular
function and promote vasoocclusion.[26]
Thrombospondin is known to bind CD47. However, although HbA and HbS
RBCs express the same amount of CD47, adhesion of TSP to HbS RBCs is
preferentially more, due to an upregulation of TSP in SCD patients
which is mediated by VLA-4.[15] Further, vWF also
induces sickle erythrocyte adhesion by interaction with endothelial
αVβ3 integrin. But, the binding of sickle RBCs to TSP was found to be
inhibited by vWF.[25] Therefore, the perturbation in
the vascular endothelium, induced by sickle RBC’s, involves complex
interactions between adhesive proteins, culminating in the VOC process
and is orchestrated by the various cytokines, a mechanism quite
different from thrombosis.
An increased risk of venous thromboembolism (VTE) is reported in non-O blood group patients,[27-30] as well as in patients with sickle cell disease. Kostner et. al[27]
reported that the odds ratio (OR) for VTE in individuals with non-O
blood groups vs. “O” blood group individuals was 2.0 (95% CI, 1.4–2.9).
After adjustment for factor VIII and VWF levels, the risk of VTE among
non-O blood group carriers was still significantly high (OR 1.5; 95%
CI, 1.0–2.2). Similar results were also reported by Tirado et al. in
2005,[28] Spiezia et al. in 2013,[29] Franchini et al. in 2014,[30] Blais et al. in 2016[31] and by Ahmed et al. in 2015[32] specifically in sickle cell trait patients.
Interestingly,
in a pilot study on consecutive SCD patients presenting with VOC, we
observed a higher incidence in “O” blood group than the non-O blood
group phenotype. Furthermore, since we know from the literature that
vWF levels in blood group “O” subjects are on an average 25% lower than
the non-O blood group subjects,[33,34] we undertook
this study, to see whether there is a relationship between blood group
“O”, vWF and TSP levels and VOC occurrence in SCD patients.
Methods
89 consecutive SCD patients (76-HbSS, 10-Hbsthalβ0, 3-Hbsthalβ+)
were prospectively enrolled in this study after informed consent and
approval by the Medical Research and Ethical Committee at the Sultan
Qaboos University Hospital. 17 patients were recruited from the
inpatient service during episodes of acute VOC’s, whereas the remaining
72 patients consented at the outpatient haematology clinics in the
steady state. vWF antigen, ristocetin cofactor activity, collagen
binding activity, vWF multimer analysis, blood group typing, CRP, HPLC,
Thrombospondin [TSP 1], TSP 2, CBC, LFT, LDH, and RFT were recorded at
enrollment. Severity and number of VOC’s /year were assessed by
stratifying these patients into 2 groups. Group 1 consists of patients
with a history of significant VOC’s, [with >4 VOC’s/year needing
inpatient care] and Group 2 consisting of SCD patients with
non-frequent-VOC’s [with <4 VOC’s/year].
Blood was obtained
by venipuncture into vacutainer tubes with Ethylene diamine tetra
acetic acid anticoagulant, 3.2% sodium citrate, and plain tubes.
Complete blood counts were performed with an electronic cell counter
(Abbott CELL-DYN® Sapphire, Abbott Diagnostics, Abbott Park, IN). A
fresh hemolysate was prepared from each sample and subjected to
cation-exchange HPLC (Bio-Rad VARIANT, Bio-Rad Laboratories, Hercules,
CA) to study the sickle phenotype. Serum was separated from a clotted
tube sample at 1,000g at 40C for 10 min and stored at -700C
for Thrombospondin [ELISA] and other biochemical assays. CRP was
estimated by rate nephelometry, in addition to the various biochemical
parameters of renal and liver function.
Whole blood samples were
collected for coagulation studies in 3.2% sodium citrate (at a ratio of
9:1 v/v) and centrifuged the same day within 2 hours of collection,
aliquoted, and stored at -700C.
Plasma activities of fibrinogen (Claus assay), coagulation assays for
prothrombin time, activated partial thromboplastin time were performed
on the same day [Dade Behring reagents]. Platelet poor plasma samples
[5 aliquots of 1 ml plasma each] were frozen to perform the vWD
phenotypic studies.
The vWF: Ag assay was performed by the Dade
Behring vWF Ag latex agglutination method for quantitative
determination of vWF Ag in human plasma by immunoturbidometry,
according to the procedure supplied by the manufacturer (Dade Behring
vWF Ag kit). The vWF: RCo was measured by platelet aggregometry using
normal lyophilized platelets with CHRONO PAR [ristocetin] for the
CHRONO-LOG aggregometer. The vWF: CBA was measured by an ELISA for the
determination of vWF function in the human plasma using Life Diagnostic
ELISA kits in duplicate, according to the manufacturer’s instructions.
Factor VIII: C levels was measured using a one-stage assay. [Dade
Behring reagents]
TSP 1 and TSP 2 concentrations in serum were
determined using the Quantikine Human Thrombospondin Immunoassay kits
[R & D systems, Minneapolis, MN]. This assay employs the
quantitative sandwich enzyme immunoassay technique and contains
NS0-expressed recombinant human Thrombospondin.
Statistical Analysis.
Data was analyzed with IBM Statistical Package for the Social Sciences
software (SPSS, version 19.0; SPSS, Chicago, Illinois, USA). Continuous
variables were expressed as mean +SD, whereas categorical variables
were expressed as numbers (percentages). Means of continuous and
categorical variables were compared using Mann-Whitney U and Fisher’s
exact tests respectively. The vWF: Ag, vWF: CBA, FVIII: C and TSP
levels showed a skewed distribution and were expressed as median values
with interquartile ranges. A p value <0.05 was considered as
statistically significant.
Results
The
mean age +SD of this SCD patient cohort was 23.8 + 6.3 (range 15 to 48
years). 50 patients were males (56%) and 17 (19%) presented with acute
VOC’s; whereas 16 (18%) (Group 1) comprised of patients with a history
of significant VOC’s [>4 VOC’s/year]. 37 patients (42%) were on
stable hydroxyurea therapy. 72 patients (81%) were enrolled in ‘‘steady
state’’ defined as “no acute illness or crisis or infection in the past
3 months” when they visited the clinic, with a prior appointment as an
outpatient. Amongst these, 37 (51%) had “O” blood group phenotype,
whereas, 35 had non-O blood group [A group -18 patients; B group -14
patients and AB group -3 patients]. None of the study participants
enrolled were on a chronic exchange transfusion program.
Table 1
summarizes the vWF parameters and TSP 1 and TSP 2 levels in the all the
steady state SCD patients [n=72]. Amongst these, in the “O” blood group
patients [n=37], the median serum TSP 1 and TSP 2 levels were
significantly higher than the non-O blood groups SCD patients [n=35]
[p<0.05, Mann-Whitney U test]. Furthermore, there was an inverse
correlation between the TSP levels and Factor VIII: C levels. The
inter-assay and intra-assay CV for thrombospondin assay were 6.3% and
5% respectively.
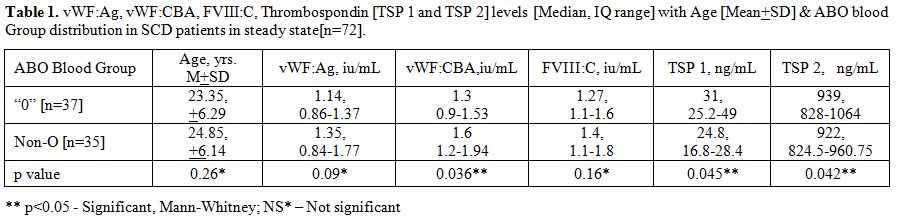 |
Table
1. vWF:Ag, vWF:CBA, FVIII:C, Thrombospondin [TSP 1 and TSP 2] levels
[Median, IQ range] with Age [Mean+SD] & ABO blood Group
distribution in SCD patients in steady state[n=72]. |
Table 2
summarizes the vWF parameters and TSP 1 and TSP 2 levels in the two
subgroups of the study participants, namely those in steady state
[n=72] and those with acute VOC’s [n=17]. In the SCD patients admitted
for VOC’s, the median serum TSP 1 and TSP 2 were significantly higher
than those in steady state SCD [p<0.05, Mann-Whitney U
test]. Furthermore, all the vWF parameters studied were significantly
lower in the painful crisis patients. The number of SCD patients with
“O” blood group was relatively higher in the painful crisis group (65%)
but was not statistically significant. However, the number of SCD
patients on HU was significantly higher in the painful crisis group
(64%), although the HbF levels were similar. The median serum TSP 1 and
TSP 2 were higher in the “O” blood group subsets comparing steady state
group and “acute” crisis group [p<0.05, Mann-Whitney U test].
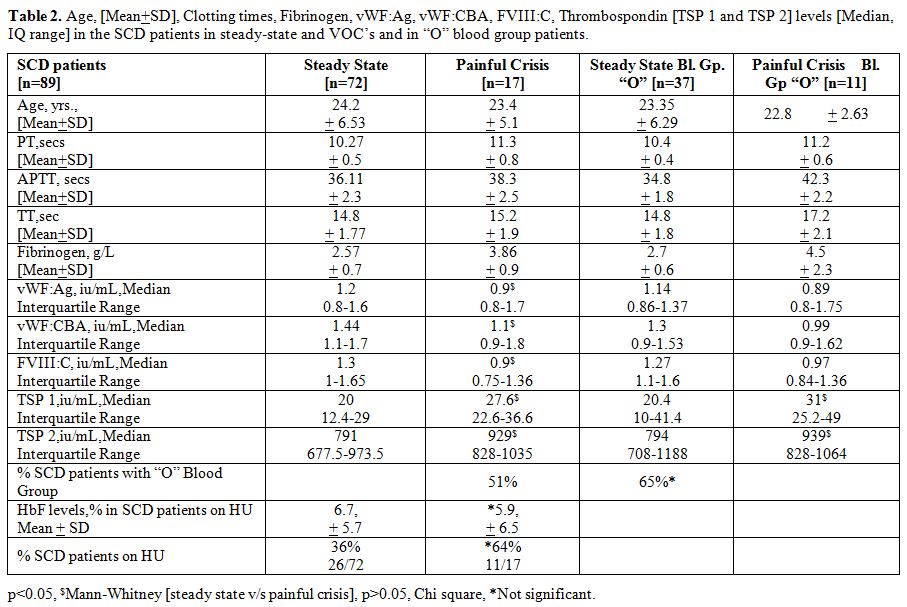 |
Table 2. Age, [Mean+SD], Clotting times,
Fibrinogen, vWF:Ag, vWF:CBA, FVIII:C, Thrombospondin [TSP 1 and TSP 2]
levels [Median, IQ range] in the SCD patients in steady-state and VOC’s
and in “O” blood group patients. |
Table 3
summarizes the vWF parameters and TSP 1 and TSP 2 levels in groups 1
and 2, namely those with a history of frequent VOC’s [n=16] and those
with infrequent VOC’s [n=73]. The median serum TSP 1 and TSP 2 were
higher in the “O” blood group subsets in both groups 1 and 2
[p<0.05, Mann-Whitney U test]. Furthermore, there was an over
representation of “O” blood group in Group 1 SCD patients (62.5%), but
this was not statistically significant. However, the number of SCD
patients on HU were significantly higher in the group 1 (68.75%),
although the HbF levels were similar.
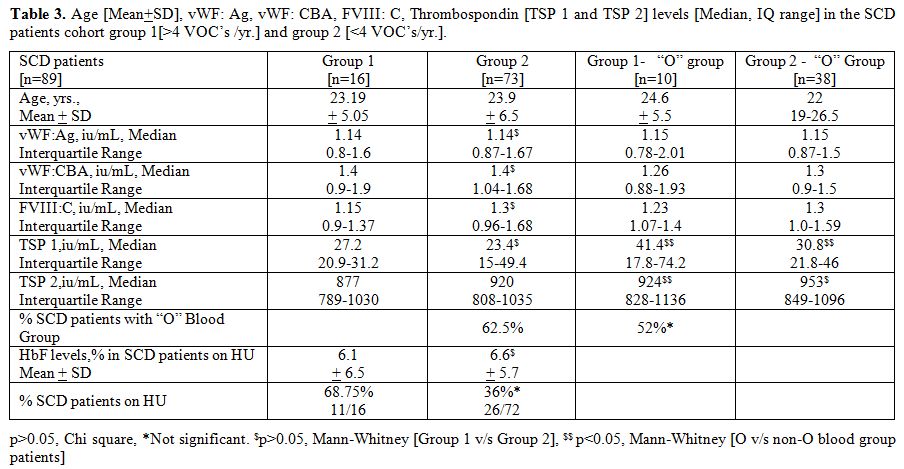 |
Table 3. Age [Mean+SD], vWF: Ag, vWF: CBA,
FVIII: C, Thrombospondin [TSP 1 and TSP 2] levels [Median, IQ range] in
the SCD patients cohort group 1[>4 VOC’s /yr.] and group 2 [<4
VOC’s/yr.]. |
Discussion
This
study documents that both serum TSP 1 and serum TSP 2 are significantly
elevated in SCD patients with VOC’s. Several investigators have
reported that TSP levels are elevated in SCD patients in crisis.[35-37] Browne et al.[35]
have reported in 1996 that plasma TSP 1 was elevated in SCD patients.
They found that TSP 1 levels were similar in normal controls and SCD
patients in steady-state, whereas these levels were significantly
elevated in SCD patients with VOC’s. They also had further documented
that the source of the raised TSP 1 in plasma was platelets, as
platelet TSP 1 levels were found depressed with a corresponding
elevation of plasma TSP levels in these SCD patients. They, therefore,
concluded that low platelet TSP levels coupled with elevated plasma TSP
levels were linked to VOC’s since these levels normalized in steady
state and became comparable to levels seen in normal controls. Further,
there was no correlation with platelet numbers and plasma TSP levels
between steady state and the vaso-occlusive crisis in these patients.
It, therefore, appears that the increased presence of markers of
platelet activation such as p-selectin, platelet factor-4, beta
thromboglobulin, soluble CD40 and platelet microparticles seen during
VOC’s is representative of the underlying inflammatory state.[38-42]
In
this study, we observed that “O” blood group was overrepresented in SCD
patients presenting with VOC’s, in comparison to the non-O blood group
SCD patients. This was documented both in patients who were frequently
admitted with VOC’s in group 1 (62.5%) as well as in patients who were
enrolled in the study as in-patients (65%). Since it is known that vWF
levels are lower in “O” blood group than non-O blood group subjects, we
investigated whether vWF levels would play a contributory role in the
occurrence of VOC’s in SCD patients. We observed that there was an
inverse relationship between TSP levels and vWF and FVIII: C during
active VOC’s, with the TSP1 and TSP2 levels being significantly
elevated (Tables 1 and 2).
Interpreting this observation in the light experimental evidence that
sickle erythrocyte adhesion to immobilized TSP is inhibited by vWF,
implies that sickle RBC adhesion is significantly influenced by the
relative concentrations of TSP and vWF in the vascular wall.[25]
Thus in “O” blood group SCD patients with a relatively lower basal vWF
levels, the relative rise in the TSP levels could promote VOC’s more
easily in comparison to the non-O blood group SCD patients.
Interestingly,
ABO blood group has been shown to have a profound influence on the
incidence of VTE, with plasma levels of vWF, being approximately 25%
higher in individuals who have non-O blood group rather than “O” blood
group.[33,34] Several case-control studies have
consistently shown that non-O blood group patients have an increased
risk for venous thrombosis[27-32] with the AB blood group having a two-fold higher risk for thrombotic vascular disease.[30]
Thus,
the important point this paper raises is that although “O” blood group
SCD patients are at a lower risk for VTE, they were actually
overrepresented amongst SCD patient with VOC’s. This implies that
mechanistic differences in pathways leading to VTE and VOC’s in SCD
patients are likely to explain the dissimilarities seen with different
underlying risk factors. In fact, VOC’s, as opposed to VTE's, is an
inflammatory condition with the clinical manifestations and
complications reflecting an interplay of several biomarkers.[38-50]
The
up-regulation of P-selectin in endothelial cells and platelets
contributes to the cell-cell interactions that are involved in the
pathogenesis of VOC’s and sickle cell-related pain. Ataga et al.,[43]
recently demonstrated that therapy with crizanlizumab, an antibody
against P-selectin, resulted in a significantly lower rate of VOC’s as
compared to placebo. In transgenic humanized SCD mice, Bennewitz et
al.,[44] recently demonstrated that microembolism of
precapillary pulmonary arteriolar vessels by neutrophil-platelet
aggregates, causing acute chest syndrome cleared following infusion of
platelet P-selectin antibody. Usefulness of both these therapeutic
approaches demonstrates the role of selectin as an important adhesive
protein that plays a significant role in the pathogenesis of sickle
VOC’s.
Selectin is important in the activation of platelets, which
is another mechanistic pathway active in sickle VOC’s. Al Najjar et
al.,[45] demonstrated that patients with SCD have
increased expression of E-selectin and P-selectin and play an important
role in the pathogenesis of VOC’s. Annarapu et al.,[46]
demonstrated that free plasma hemoglobin present following
intravascular hemolysis in SCD binds to glycoprotein 1bα, inducing the
activation of platelets. Wu et al.,[47] in a double
blind, randomized study showed that prasugrel, a third-generation
thienopyridine, was able to decrease platelet activation biomarkers and
reduce sickle-cell VOC pain as compared to placebo. Although it is
believed that platelets mediate intercellular adhesion during sickle
cell VOC’s, Heeney et al,[48] in an international
multicenter study utilizing prasugrel, failed to show a statistically
significant reduction in VOC’s, although there was a trend to show a
reduction in the VOC pain.
Low molecular weight heparins (LMWH) have been used to control the hypercoagulability associated with sickle cell VOC’s.[49-50] In a randomized study using Tinzaparin, Qari et al.,[49]
showed reduced severity and duration of acute crisis in sickle cell
anemia. However, well-designed placebo-controlled studies with
different LMWH, and enrolling participants with different genotypes of
sickle cell disease are lacking.[50] Telen et al.[51]
demonstrated that sevuparin, a heparin-derived polysaccharide, reduced
sickle cell related VOC’s. The efficacy of sevuparin is believed to be
due to its anti-adhesive properties, as it binds to P-and L-selectins,
TSP, fibronectin, and VWF, all of which are involved in the sickle cell
VOC’s.
Lastly, it has also been reported that a high level of
extracellular hemoglobin plays an important role in SCD patients since
nitric oxide (NO) quenching mechanism are compromised.[52,53]
The free hemoglobin binds not only to vWF multimers but also with
ADAMTS-13, leading to an acquired ADAMTS-13 deficiency, blocking
appropriate proteolysis of vWF, causing the accumulation of ultra-large
vWF multimers. However, using real-time fluorescence intravital
microscopy, Barazia et al.[54] showed that plasma nitric oxide levels could be normalized by using hydroxyurea therapy.
Overall,
therefore, it is quite apparent that SCD is actually a well-recognized
state of chronic indolent inflammation and there indeed exist several
lines of evidence demonstrating the mechanistic differences in VOC
pathways as against VTE pathways. Adhesive proteins like selectins and
TSP decelerate sickle red cells and the platelet-leukocytes
interactions in the circulation, facilitating endothelial adhesion and
other cell-cell interactions, ultimately leading to vascular occlusion
in sickle VOC’s. However, the occurrence of VTE depends on its
predisposing risk factors.
The major drawback of this study is
the small number of evaluable patients. Although the study
prospectively enrolled consecutive patients for almost 2 years, we were
able to get only a total of 89 SCD patients. Nevertheless, our data are
valuable as they show observations in an ethnic SCD population that
have not been reported before.
Conclusions
Abnormal
adhesive interactions between sickle erythrocytes and vascular
endothelial cells and/or subendothelial matrix play a significant role
in the initiation of sickle VOC’s. Selectins, TSP and vWF are important
mediators of the adhesive interactions between sickle erythrocytes and
the blood vessel wall. Our study showed an inverse relation between TSP
and vWF levels, in blood group “O” SCD patients with elevated TSP
levels during active VOC”s.
Acknowledgements
We wish to thank the
Hospital Administration for allowing the use of hospital data. This
work was supported by a research grant no. IG/MED/HAEM/10/02.
References
- Luzzatto L. Sickle cell anemia in tropical Africa. Clin Hematol 1981; 3: 757–784.

- Serjeant GR. Sickle cell disease. Oxford, England: Oxford University Press; 1992.
- Bassiouny
MR, Lamki Z, Elbanna N, Shah, WM, White JM. Sickle Cell Disease, A
Clinico-Epidemiological Study from Oman. Bahrain Med Bull
1995;17:101–104. http://www.bahrainmedicalbulletin.com/september_1995/sickle-cell_anaemia.pdf
- Daar
S, Hussain HM, Gravell D, Nagel RL, Krishnamoorthy R. Genetic
epidemiology of HbS in Oman: multicentric origin for the betaS gene. Am
J Hematol 2000; 64: 39–46. https://doi.org/10.1002/(sici)1096-8652(200005)64:1<39::aid-ajh7>3.3.co;2-r
- Makis AC, Hatzimichael EC, Bourantes KL. The role of cytokines in sickle cell disease. Ann Hematol 2000; 79: 407–413. https://doi.org/10.1007/s002770000173
- Belcher
JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes
in sickle cell disease: potential role in the activation of vascular
endothelium and vaso-occlusion, Blood 2000;96:2451–2459.
- Brittain
JE, Mlinar KJ, Anderson CS, Orringer EP, Parise LV. Activation of
sickle red blood cell adhesion via integrin-associated protein/CD
47-induced signal transduction. J Clin Invest 2001; 107: 1555–1562. https://doi.org/10.1172/jci10817
- Haynes
J, Obiako B. Activated polymorphonuclear cells increase sickle red
blood cell retention in lung: role of phospholipids. Am J Physiol Heart
Circ Physiol 2002; 282: H122–H130.
- Wun
T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated
monocytes and platelet–monocyte aggregates in patients with sickle cell
disease. Clin Lab Haematol 2002;24:81–88. https://doi.org/10.1046/j.1365-2257.2002.t01-1-00433.x
- Pathare
AV, Al Kindi S, Alnaqdy AA, Mohite U, Hiwase D, Dennison D, Daar D,
Knox-Macaulay HH. Cytokine profile of sickle cell disease patients from
Oman in acute crisis vs steady state. Blood 2002; 100: 28b [abstract
3565].
- Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest 1997; 99: 2561–2564. https://doi.org/10.1172/jci119442
- Bookchin RM, Lew VL. Pathophysiology of sickle cell anemia. Hematol Oncol Clin North Am, 1996; 10:1241-1253. https://doi.org/10.1016/s0889-8588(05)70397-x
- Rodgers GP. Overview of pathophysiology and rationale for treatment of sickle cell anemia. Semin Hematol 1997; 34:2-7.
- Ballas SK, Mohandas N. Pathophysiology of vaso-occlusion. Hematol Oncol Clin North Am 1996; 10:1221-1239. https://doi.org/10.1016/s0889-8588(05)70396-8
- C.C.
Joneckis, R.L. Ackley, E.P. Orringer, E.A. Wayner, L.V. Parise Integrin
alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating
reticulocytes in sickle cell anemia. Blood,1993; 82: 3548–3555.
- R.A.
Swerlick, J.R. Eckman, A. Kumar, M. Jeitler, T.M. Wick, Alpha 4 beta
1-integrin expression on sickle reticulocytes: vascular cell adhesion
molecule-1-dependent binding to endothelium, Blood, 1993; 82, 1891–1899
- J.E.
Brittain, K.J. Mlinar, C.S. Anderson, E.P. Orringer, L.V. Parise,
Integrin-associated protein is an adhesion receptor on sickle red blood
cells for immobilized thrombospondin, Blood, 2001; 97, 2159–2164. https://doi.org/10.1182/blood.v97.7.2159
- Pathare A, AlKindi S, Daar S, and Dennison D, Cytokines in Sickle Cell Disease, Hematology, 2003, Vol.8(5) Oct. Pg-329-337. https://doi.org/10.1080/10245330310001604719
- Mosseri
M, Bartlett-Pandite AN, Wenc K, Isner MH, Weinstein R. Inhibition of
endothelium-dependent vasorelaxation by sickle erythrocytes. Am Heart J
1993; 126:338-346. https://doi.org/10.1016/0002-8703(93)91049-k
- Hebbel RP, Vercelotti GM. The endothelial biology of sickle cell disease. J Lab Clin Med 1997; 129:288-293. https://doi.org/10.1016/s0022-2143(97)90176-1
- Hebbel
RP, Yamada O, Moldow CF, Jacob HS. White JG, Eaton JW. Abnormal
adherence of sickle erythrocytes to cultured vascular endothelium:
Possible mechanism for microvascular occlusion in sickle cell disease.
J Clin Invest, 1980; 65:154. https://doi.org/10.1172/jci109646
- Hebbel
RP, Mohandas N. Sickle cell adherence. In Embury SH, Hebbel RP,
Mohandas N, Steinberg MH (Eds): Sickle cell disease: Basic principles
and clinical practice. New York, NY, Raven, 1994, p 217.
- Kaul
DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of
sickle cell adhesion to vascular endothelium in shear flow conditions:
Pathophysiological implications. Proc Natl Acad Sci USA, 1989; 86:3356.
https://doi.org/10.1073/pnas.86.9.3356
- Barabino
GA, McIntire LV, Eskin SG, Sears DA, Udden M: Endothelial cell
interactions with sickle cell, sickle trait, mechanically injured, and
normal erythrocytes under controlled flow. Blood, 1987; 70:152.
- Barabino
GA, Wise RJ, Woodbury VA, Zhang B, Bridges KA, Hebbel RP, Lawler J,
Ewenstein BM. Inhibition of sickle erythrocyte adhesion to immobilized
thrombospondin by von Willebrand factor under dynamic flow conditions.
Blood, 1997, 89:2560.
- Camus
SM, Gausserès B, Bonnin P, Loufrani L, Grimaud L, Charue D, De Moraes
JA, Renard JM, Tedgui A, Boulanger CM, Tharaux PL, Blanc-Brude OP.
Erythrocyte microparticles can induce kidney vaso-occlusions in a
murine model of sickle cell disease. Blood. 2012 Dec 13;
120(25):5050-8. https://doi.org/10.1182/blood-2012-02-413138.
- Koster
T, Blann AD, Briet E, Vandenbrouche JP, & Rosendaal FR. Role of
clotting factor VIII in effect of von Willebrand factor on occurrence
of deep-vein thrombosis. Lancet. 1995; 345:152–5. https://doi.org/10.1016/s0140-6736(95)90166-3
- Tirado
I, Mateo J, Soria JM, Oliver A, Martinez-Sanchez E, Vallve C, Borrell
M, Urrutia T & Fontcuberta J. The ABO blood group genotype and
factor VIII levels is independent risk factors for venous
thromboembolism. Thromb Haemost. 2005; 93: 468–74. https://doi.org/10.1160/th04-04-0251
- Spiezia
L, Campello E, Bon M, Tison T, Milan M, Simioni P & Prandoni P. ABO
blood groups and the risk of venous thrombosis in patients with
inherited thrombophilia. Blood Transfus. 2013; 11: 250–3. https://doi.org/ 10.2450/2012.0060-12
- Franchini M, Mannucci PM. ABO blood group and thrombotic vascular disease. Thromb Haemost. 2014 Dec;112(6):1103-9. https://doi.org/10.1160/th14-05-0457
- Blais
C, Germain M, Delage G, Grégoire Y. The association between blood group
and the risk of vascular disease in Quebec blood donors. Blood
Transfus. 2016 Sep;14(5):455-9. https://doi.org/10.2450/2016.0303-15
- Ahmed
SG, Kagu MB, Ibrahim UA, Bukar AA. Impact of sickle cell trait on the
thrombotic risk associated with non-O blood groups in northern Nigeria.
Blood Transfus. 2015 Oct;13(4):639-43. https://doi.org/10.2450/2015.0335-14
- Gill
JC, Endres-Brooks J, Bauer PJ, Marks WJ Jr, & Montgomery RR. The
effect of ABO blood group on the diagnosis of von Willebrand disease.
Blood. 1987;69:1691–5.
- Jenkins
PV, O'Donnell JS. ABO blood group determines plasma von Willebrand
factor levels: a biologic function after all? Transfusion. 2006
Oct;46(10):1836-44.
- Browne
PV, Mosher DF, Steinberg MH, Hebbel RP. Disturbance of plasma and
platelet thrombospondin levels in sickle cell disease. Am J Hematol
1996; 51: 296-301. https://doi.org/10.1002/(sici)1096-8652(199604)51:4<296::aid-ajh8>3.0.co;2-r
- Novelli
EM, Kato GJ, Ragni MV, Zhang Y, Hildesheim ME, Nouraie M, Barge S,
Meyer MP, Hassett AC, Gordeuk VR, Gladwin MT, Isenberg JS. Plasma
thrombospondin-1 is increased during acute sickle cell vaso-occlusive
events and associated with acute chest syndrome, hydroxyurea therapy,
and lower hemolytic rates. American Journal of Hematology 2012; 87 (3):
326-330. https://doi.org/10.1002/ajh.22274
- Novelli
EM, Kato GJ, , Hildesheim ME, Barge S, Meyer MP, Lozier J, Hassett AC,
Ragni1 MV, Isenberg JS and, Gladwin MT. Thrombospondin-1 inhibits
ADAMTS13 activity in sickle cell disease. Haematologica November 1,
2013; 98 (11): e132-e134. https://doi.org/10.3324/haematol.2013.092635
- Hagag
AA, Elmashad G, Abd El-Lateef AE. Clinical significance of assessment
of thrombospondin and placenta growth factor levels in patients with
sickle cell anemia: two centers Egyptian studies. Mediterr J Hematol
Infect Dis. 2014 Jul 1; 6(1):e2014044. https://doi.org/10.4084/mjhid.2014.044
- Mohan
JS, Lip GY, Bareford D, Blann AD. Platelet P-selectin and platelet
mass, volume and component in sickle cell disease: Relationship to
genotype. Thromb Res 2006; 117: 623–629. https://doi.org/10.1016/j.thromres.2005.05.010
- Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med 2001; 137: 398–407. https://doi.org/10.1067/mlc.2001.115450
- Wun
T, Paglieroni T, Rangaswami A, Franklin PH, Welborn J, Cheung A, Tablin
F. Platelet activation in patients with sickle cell disease. Br J
Haematol 1998; 100: 741–749. https://doi.org/10.1046/j.1365-2141.1998.00627.x
- Lee
SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active
CD40 ligand is elevated in sickle cell anemia: Potential role for
platelet-mediated inflammation. Arterioscler Thromb Vasc Biol 2006; 26:
1626–1631. https://doi.org/10.1161/01.atv.0000220374.00602.a2
- Ataga
KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH,
Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith
WR, Rollins SA, Stocker JW, Rother RP. Crizanlizumab for the Prevention
of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017 Feb
2;376(5):429-439. https://doi.org/10.1056/NEJMoa1611770
- Bennewitz
MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, Kato GJ, Gladwin
MT, Sundd P. Lung vaso-occlusion in sickle cell disease mediated by
arteriolar neutrophil-platelet microemboli. JCI Insight. 2017 Jan
12;2(1):e89761. https://doi.org/10.1172/jci.insight.89761
- Al
Najjar S, Adam S, Ahmed N, Qari M. Markers of endothelial dysfunction
and leucocyte activation in Saudi and non-Saudi haplotypes of sickle
cell disease. Ann Hematol. 2017 Jan;96(1):141-146. https://doi.org/10.1007/s00277-016-2823-7
- Annarapu
GK, Singhal R, Gupta A, Chawla S, Batra H, Seth T, Guchhait P. HbS
Binding to GP1bα Activates Platelets in Sickle Cell Disease. PLoS One.
2016 Dec 9; 11(12):e0167899. https://doi.org/10.1371/journal.pone.0167899
- Wun
T, Soulieres D, Frelinger AL, Krishnamurti L, Novelli EM, Kutlar A,
Ataga KI, Knupp CL, McMahon LE, Strouse JJ, Zhou C, Heath LE, Nwachuku
CE, Jakubowski JA, Riesmeyer JS, Winters KJ. A double-blind,
randomized, multicenter phase 2 study of prasugrel versus placebo in
adult patients with sickle cell disease. J Hematol Oncol. 2013 Feb
17;6:17. https://doi.org/10.1186/1756-8722-6-17
- Heeney
MM, Hoppe CC, Abboud MR, Inusa B, Kanter J, Ogutu B, Brown PB, Heath
LE, Jakubowski JA, Zhou C, Zamoryakhin D, Agbenyega T, Colombatti R,
Hassab HM, Nduba VN, Oyieko JN, Robitaille N, Segbefia CI, Rees DC;
DOVE Investigators.. A Multinational Trial of Prasugrel for Sickle Cell
Vaso-Occlusive Events. N Engl J Med. 2016 Feb 18;374(7):625-35. https://doi.org/10.1056/NEJMoa1512021
- Qari
MH, Aljaouni SK, Alardawi MS, Fatani H, Alsayes FM, Zografos P, Alsaigh
M, Alalfi A, Alamin M, Gadi A, Mousa SA. Reduction of painful
vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a
double-blind randomized trial. Thromb Haemost. 2007 Aug;98(2):392-6. https://doi.org/10.1160/th06-12-0718
- van
Zuuren EJ, Fedorowicz Z. Low-molecular-weight heparins for managing
vaso-occlusive crises in people with sickle cell disease. Cochrane
Database Syst Rev. 2015 Dec 18;(12):CD010155. https://doi.org/10.1002/14651858
- Telen
MJ, Batchvarova M, Shan S, Bovee-Geurts PH, Zennadi R, Leitgeb A, Brock
R, Lindgren M. Sevuparin binds to multiple adhesive ligands and reduces
sickle red blood cell-induced vaso-occlusion. Br J Haematol. 2016
Dec;175(5):935-948. https://doi.org/10.1111/bjh.14303
- Zhou
Z, Han H, Cruz MA, López JA, Dong JF, Guchhait P. Haemoglobin blocks
von Willebrand factor proteolysis by ADAMTS-13: a mechanism associated
with sickle cell disease. Thromb Haemost. 2009 Jun;101(6):1070-77. https://doi.org/10.1160/th08-10-0677
- Zhou
Z, Guchhait P. Extracellular hemoglobin regulation of von Willebrand
factor activity in plasma of patients with sickle cell disease. US
Oncol Hematol 2011; 7:150–152. https://doi.org/10.17925/ohr.2011.07.2.150
- Barazia
A, Li J, Kim K, Shabrani N, Cho J. Hydroxyurea with AKT2 inhibition
decreases vaso-occlusive events in sickle cell disease mice. Blood.
2015 Nov 26;126(22):2511-7. https://doi.org/10.1182/blood-2015-02-626234
[TOP]