Jerome Guison1, Gilles Blaison1, Oana Stoica1, Remy Hurstel2, Marie Favier3 and Remy Favier4
1 Service
de médecine interne et maladies infectieuses, Centre Haut-Rhinois de
compétence des maladies systémiques et auto-immunes rares, Hôpital
Pasteur, Hôpitaux civils de Colmar, 39 avenue de la liberté, 68024
COLMAR.
2 Laboratoire d’hématologie et d’hémostase, Hôpital Pasteur, Hôpitaux civils de Colmar, 39 avenue de la liberté, 68024 COLMAR.
3 Faculté de médecine, INRA/UMR 1260; 27 boulevard J. Moulin, 13385 MARSEILLE.
4
Service d’hématologie biologique, Centre de référence des pathologies
plaquettaires, Assistance Publique-Hôpitaux de Paris, Hôpital Armand
Trousseau, 26 avenue du Dr Netter, 75012 PARIS
Corresponding
author: Jérôme Guison, M.D. Service de
médecine interne et maladies infectieuses, Centre Haut-Rhinois de
compétence des maladies systémiques et auto-immunes rares. Hôpital
Pasteur, Hôpitaux civils de Colmar, 39 avenue de la liberté, 68024
COLMAR. Tel: +33 (0) 389124123 - Fax: +33 (0) 389124691. E-mail:
jerome.guison@ch-colmar.fr
Published: June 16, 2017
Received: February 17, 2017
Accepted: May 15, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017038 DOI
10.4084/MJHID.2017.038
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Venous
thrombosis affecting thrombocytopenic patients is challenging. We
report the case of a woman affected by deep vein thrombosis and
pulmonary embolism in a thrombocytopenic context leading to the
discovery of a heterozygous mutation in the gene encoding ankyrin
repeat domain 26 (ANKRD26) associated with a heterozygous factor V (FV)
Leiden mutation. This woman was diagnosed with lower-limb deep vein
thrombosis complicated by pulmonary embolism. Severe thrombocytopenia
was observed. The genetic study evidenced a heterozygous FV Leiden
mutation. Molecular study sequencing was performed after learning that
her family had a history of thrombocytopenia. Previously described
heterozygous mutation c-127C>A in the 5′untranslated region (5′UTR)
of the ANKRD26 gene was detected in the patient, her aunt, and her
grandmother. ANKRD26-related thrombocytopenia and thrombosis are rare.
This is, to our knowledge, the first case reported in the medical
literature. This mutation should be screened in patients with a family
history of thrombocytopenia.
|
Case Report
A 20-year-old woman (Figure 1, III-3)
was admitted to our department after being diagnosed with spontaneous
left lower limb deep vein thrombosis (DVT) located in the external
iliac vein. She had no recent history of travel, limb surgery, or
smoking habits. Her medical background consisted of one episode of
infectious pneumopathy (1 year before this case) complicated with
pachypleuritis, an upper humeral extremity fracture, and migraines. No
thrombosis episodes were known in the family. Her medication consisted
of a combined oral contraceptive pill taken for two years
(levonorgestrel 0.150 mg/ethinyl estradiol 0.030 mg). Except for an
edematous limb, the patient was asymptomatic. Laboratory investigations
(Sysmex XE-5000 and XE-2100) revealed the following: hemoglobin, 13.4
g/dL; microcytic red blood cells (mean corpuscular volume, MCV), 76µ3;
increased white blood cell (WBC) count, 15.6 G/L; and low platelet
count, 35 G/L. Mean platelet volume (MPV) was normal (11 fL; laboratory
rates: 7.2–11.1 fL). Prothrombin time was 12.6 s (P/C ratio, 1.14) and
activated partial thromboplastin time, 28.5 s (P/C ratio, 0.91). The
iron stock was normal, as were protein electrophoresis, antibodies, and
other biological parameters. Arterial gasometry showed a normal pH
(7.43) and a shunt effect. Electrocardiogram and transthoracic
echocardiography (left ventricular ejection fraction, 67%) were normal.
Functional respiratory investigations were slightly modified. Bone
marrow study was not performed. We retrieved the patient’s
hematological parameters performed one year before during the
infectious episode: platelet count was then low at 134 G/L with normal
MPV (10.1 fL), red blood cells were microcytic (MCV: 79.8 µ3)
with anemia (hemoglobin, 9.5 g/dL). WBC count was increased to 20 G/L
with a high level of C-reactive protein at 332 mg/L and procalcitonin
at 1.25 ng/mL.
Taking into account the low platelet count, we used
Fondaparinux (7.5 mg/day) for four days as an anticoagulant treatment,
relayed by Rivaroxaban 30 mg/day for three weeks, then 20 mg/day for
six months. Although the patient was asymptomatic, pulmonary
scintigraphy showed a massive bilateral pulmonary embolism.
Thrombophilia testing revealed a heterozygous R506Q FV Leiden mutation
with no other abnormalities. Lupus anticoagulant, anti-β2GPI
antibodies, and anticardiolipin were negative. Oxygen (2L/min) was
provided for a total of 6 days. Finally, the diagnosis of inherited
thrombocytopenia was suspected because there was a family history of
unexplained thrombocytopenia with at least six members known to be
thrombocytopenic (Figure 1): her two sisters (Figure 1; III-1 and III-2); her father (Figure 1; II-4); two aunts (Figure 1; II-2 and II-3), and her grandmother (Figure 1; I-2).
About the grandmother, we managed to retrieve the platelet count in
2015; they were at 32 G/L at the time. Interestingly, she suffered from
6 pulmonary embolisms (and was treated from 1995 with anti-vitamin K
anticoagulant), but never suffered from any bleeding complications.
Sanger
sequencing of the 5′UTR part of the ANKRD26 gene demonstrated a
previously described heterozygous c-127 C>A mutation. Knowing that
result, we decided to conduct familial genetic investigations.
Interestingly, the patient’s grandmother was affected by refractory
cytopenia with multilineage dysplasia. We evidenced two family members
positive for this mutation: the maternal grandmother (Figure 1; I-2) and one aunt (Figure 1; II-2).
Nine days after admission, the patient was discharged. Six months after
this thromboembolic event, anticoagulant treatment was stopped with no
further complications. The patient was lost to follow-up after this
last medical consultation, due to a job transfer. This observation is,
to our knowledge, the first case of a pulmonary thromboembolic event in
a patient with an ANKR26 inherited mutation and a factor V Leiden
heterozygous mutation.
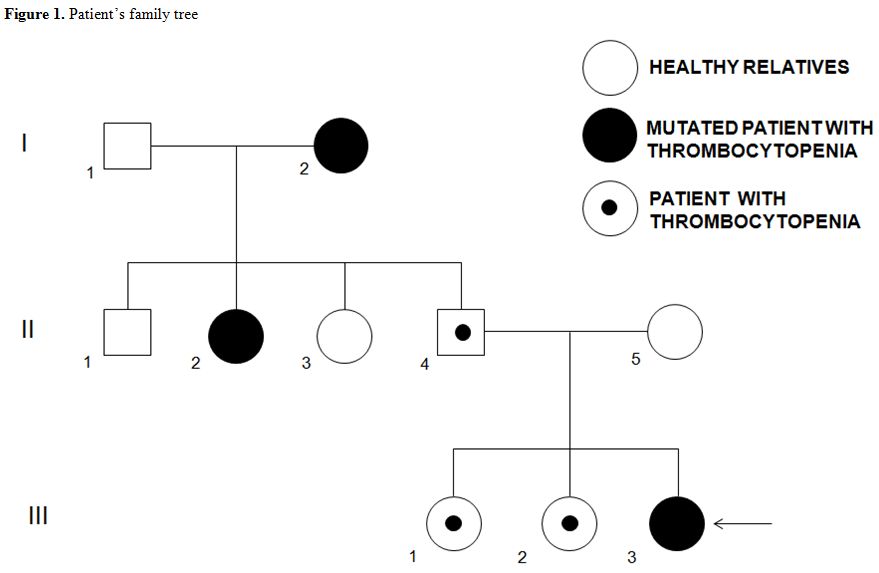 |
Figure 1. Patient’s family tree |
Discussion
Thrombosis
associated with inherited platelet disorders (IPDs) is rare. Girolami
et al reviewed three different inherited platelet disorders with
thrombotic events: two cases of inherited thrombocytopenia with
increased MPV, i.e., MYH9 (Myosin Heavy Chain 9) (OMIM 155100) and
Bernard-Soulier syndromes (BSS) (OMIM 231200), and a platelet function
disorder with a normal MPV in Glanzmann thrombasthenia (GT, OMIM
273800).[1] In the majority of these cases, patients
developed thrombotic events after identification of the inherited
platelet disorders. To our knowledge, the present report is the first
in which a venous thrombotic event resulted in the discovery of
inherited thrombocytopenia (IT): ANKRD26-related thrombocytopenia (OMIM
18800). Nevertheless, these four platelet disorders differ from two
other prothrombotic dysfunctional platelet disorders: the Wien-Penzing
defect[2] and sticky platelet syndrome (SPS,)[3] characterized by the occurrence of venous or arterial thrombosis that usually reveals these platelet dysfunctions.
Thrombotic events described in the literature are mostly arterial: nine MYH9 patients[1,4–5]
and three BSS patients developed myocardial infarction, coronary
arterial disease, and pons infarction stroke. In contrast, ten
Glanzmann patients developed venous thrombosis,[1,6] mas did two MYH9 patients.[1]
Traditional risk factors (long flight immobilization, old age, surgery,
treatment) associated with venous thrombosis are evidenced in 36% of
the cases, other risk factors (V Leiden or JAK2 mutations) in 28%,
unknown or undetected risk factors in 36%. In arterial thrombosis
events, associated traditional risk factors (HTA, atrial fibrillation,
hyperlipidemia, elevated levels of homocysteinemia or cholesterol,
atherosclerosis, smoking) are present in 67% of the cases and
undetected or absent in 33%. As in our patient, heterozygous FV
mutation was detected in three Glanzmann patients with recurrent deep
venous thrombosis[7,8] and a JAK2V617F mutation in an MYH9 patient with portal vein thrombosis.[1]
The
discrepancy between the macrothrombocytopenia group and Glanzmann
patients for thrombotic events remains unexplained because thrombosis
may occur whether a functional platelet defect is present (BSS,
Glanzmann) or not (MYH9, ANKRD26). In addition, functional defects in
Glanzmann and BSS patients are distinct: it has been suggested that the
defective binding of Von Willebrand factor (alteration present in BSS)
can protect from venous thrombosis, whereas defective binding of
fibrinogen (seen in Glanzmann) protects more from arterial thrombosis.[7]
This hypothesis could not be totally applied to MYH9 syndrome or our
case because these two IPDs have no functional defect. A recent,
interesting report found no difference in thrombin potential generation
in MYH9 patients with or without arterial thrombotic events, indicating
that other factors than the low platelet count might have contributed
to the thrombosis.[5] However, few patients were
tested in this study. One should also keep in mind not only that few
patients who developed thrombosis have been described, but also that
the frequency of the different forms of IT is variable. We cannot
exclude that in the present report, the risk factors themselves induced
venous thrombosis and that the association made herein might be
fortuitous.
The IT type diagnosed in this report should also be
described. In our patient’s family, thrombocytopenia was a
non-syndromic autosomal dominant form with an average MPV. In this
disorder, morphologically platelets appeared normal but could be
macrocytic,[9,10] sometimes demonstrated with optical
microscopy. Electron microscopy studies identified the presence of
particulate cytoplasmic structures in ANKRD26 platelets and
megakaryocytes of patients with mutations reflecting dysfunctional
proteasome pathways.[11] In the group of IT patients
with normal MPV, three genes must be sequenced first and foremost: the
ANKRD26 gene (OMIM 188000), the RUNX1 gene mutated in the familial
thrombocytopenic disorder with predisposition to myeloid leukemia (OMIM
601399), and a recently described gene, ETV6, which confers a
predisposition to lymphoid leukemia and solid tumors (OMIM 6000618;).[12]
Platelet counts of different ANKRD26 patients published to date vary
between 8 and 107 G/L, two patients having normal values.[9,10] There are reports of transient normalization of platelet counts in the setting of an acute infection,[10]
as observed in our patient, probably explained by the thrombopoietin
level increase, usually found in inflammatory conditions.[13]
The bleeding syndrome is usually moderate, and numerous patients have
undergone surgeries without platelet support and bleeding. No bone
marrow analysis was performed for this diagnosis, but when previously
performed, a dysmegakaryopoiesis is described, which contributes to the
thrombocytopenia mechanism.[10] Even if severe
bleeding affects only a minority of patients, recent studies have
pointed out the importance of IT genetic diagnosis.[12]
The
majority of the mutations identified are single nucleotide
substitutions in the 5′UTR part of the ANKRD26 gene: the most frequent
mutations described were c-127 A>T, c-128G>A, and c-134G>A.[9,10,14] One missense mutation in the coding region of the gene was also reported in one family.[15]
These mutations might result in the loss of two binding transcription
factors that inhibit ANKRD26 expression in normal conditions and induce
abnormal persistent activation of the ERK/MAP pathway, leading to the
impaired pro-platelet formation and dysmegakaryopoiesis.[16]
Today,
it is well established that some ITs are characterized by increased
risk of acquiring additional disorders over time that are much more
relevant for patients than thrombocytopenia itself. Patients with
ANKRD26-related thrombocytopenia have a propensity to develop myeloid
malignancies.[17,18] Therefore, recognizing such
patients is essential to provide genetic counseling and personalize
follow-up, especially if hematological malignancies occur.
Conclusions
This
first report adds to previous observations and confirms that platelet
defects do not protect from venous thrombosis, in particular, the
ANKRD26 familial thrombocytopenia. It remains unexplained why
thrombotic events appear in some inherited platelet disorders, but if
we want to explain this association, this would require larger studies
with more patients having rare platelet disorders which are not
possible up to date. International guidelines for clinical and
biological follow-up of these patients with inherited platelet
disorders predisposing patients to hematological malignancies are
therefore needed.
Authorship details
JG and RF conducted the literature review, drafted the manuscript, and made the figures.
All the authors revised the manuscript and read and approved its final version.
Acknowledgements
The authors would like to thank Ms. Christine Nguyen for her technical assistance.
References
- Girolami A, Sambado L, Bonamigo E, Vettore S,
Lombardi AM. Occurrence of thrombosis in congenital thrombocytopenic
disorders: a critical annotation of the literature. Blood Coagul
Fibrinolysis 2013 Jan;24(1):18-22. https://doi.org/10.1097/MBC.0b013e3283597634

- Sinzinger
H, Kaliman J, O'Grady J. Platelet lipoxygenase defect (Wien-Penzing
defect) in two patients with myocardial infaction. Am J Hematol
1991;36(3):202-205. https://doi.org/10.1002/ajh.2830360308 PMid:1899965
- Kubisz
P, Stanciakova L, Stasko J, Dobrotova M, Sterenova M, Ivankova J, Holly
P. The sticky platelet syndrome: an important cause of life-threatening
thrombotic complications. Expert Rev Hematol 2016 Jan;9(1):21-35. Epub
2015 Dec 9. https://doi.org/10.1586/17474086.2016.1121095
- Althaus
K, Greinacher A. MYH-9 related platelet disorders. Strategies for
management and diagnosis. Transfus Med Hemother 2010;37(5):260-267.
Epub 2010 Sep 15. https://doi.org/10.1159/000320335 PMid:21113248 PMCid:PMC2980510
- Zetterberg
E, CarissonAlle MS, Najm J, Greinacher A. Thrombin generation in two
families with MYH9 related platelet disorder. Platelets
2016;27(3):264-7. Epub 2015 Aug 6 https://doi.org/10.3109/09537104.2015.1064882
- Nurden
AT, Fiore M, Nurden P, Pilois X. Glanzmann thrombasthenia: a review of
ITGA2B and ITGB3 defects with emphasis on variants, phenotypic
variability and animal models. Blood 2011 Dec 1;118(23):5996-6005. Epub
2011 Sep 13. https://doi.org/10.1182/blood-2011-07-365635
- Ten
Cate H, Brandjes DPM, Smits PHM, Van Mourik JA. The role of platelets
in venous thrombosis: a patient with glanzmann's thrombasthenia and a
factor V Leiden mutation suffering from deep venous thrombosis. J
ThrombHaem2003; 1:394-395. https://doi.org/10.1046/j.1538-7836.2003.00041.x PMid:12871523
- Rezende
SM. Secondary prohylaxis with warfarin for recurrent thrombosis in a
patient with Glanzmannthrombastenia and F5 G1691A. Br J Haematol
2012;156:144-145. https://doi.org/10.1111/j.1365-2141.2011.08821.x PMid:21848888
- Pippucci
T, Savoia A, Perrotta S, Pujol-Noix N, Noris P, Castegnaro G, et al.
Mutations in the 5' UTR of ANKRD26, the ankyrin repeat domain 26 gene,
cause an autosomal-dominant form of inherited thrombocytopenia, THC2.
Am J Hum Genet 2011;88(1):115-120. https://doi.org/10.1016/j.ajhg.2010.12.006 PMid:21211618 PMCid:PMC3014357
- Noris
P, Perrotta S, Seri M, Pecci A, Gnan C, Loffredo G et et al. Mutations
in ANKRD26 are responsible for a frequent form of inherited
thrombocytopenia analysis of 78 patients from 21 families. Blood
2011;117(34):6673-6680. https://doi.org/10.1182/blood-2011-02-336537 PMid:21467542
- Necchi
V, Balduini A, Noris P, Barozzi S, Sommi P, Di Buduo C et al.
Ubiquitin/proteasome rich particulate cytoplasmic structures (PaCs) in
the platelets and megakaryocytes of ANKRD26-related thrombocytopenia.
Thromb Haemost 2013;109 :263-71. https://doi.org/10.1160/TH12-07-0497 PMid:23223974
- Balduini CL, Savoia A, Seri M. Inherited thrombocytopenias frequently diagnosed in adults. J Thromb Haemost 2013;11(6):1006-19. https://doi.org/10.1111/jth.12196 PMid:23510089
- Cerutti
A, Custodi P, Duranti M, Cazzola M, Balduini CL. Circulating
thrombopoietin in reactive conditions behaves like an acute phase
reactant. Clin Lab Haematol 1999;21(4):271-275. https://doi.org/10.1046/j.1365-2257.1999.00226.x PMid:10583330
- Marquez
R, Hantel A, Lorentz R, Neistadt B, Wong J, Churpek JE et al. A new
family with a germline ANKRD26 mutation and predisposition to myeloid
malignancies. Leuk Lymphoma 2014;55:2945-6. https://doi.org/10.3109/10428194.2014.903476 PMid:24628296 PMCid:PMC4206674
- Al
Daama SA, Housawi Y, Dridi W, Sager M, Otieno FG et al. A misssense
mutation in ANKRD26 segregates with thrombocytopenia. Blood
2013;122:481-2. https://doi.org/10.1182/blood-2013-03-489344 PMid:23869080
- Bluteau
D, Balduini A, Balayn N, Currao M, Nurden P, Deswarte C et al.
Thrombocytopenia associated mutations in the ANKRD26 regulatory region
induce MAPK hyperactivation. J Clin Invest 2014;124(2):580-91. https://doi.org/10.1172/JCI71861 PMid:24430186 PMCid:PMC3904625
- Noris
P, Favier R, Alessi MC, Geddis AE, Kunishima S, Heller PG, et al.
ANKRD26-related thrombocytopenia and myeloid malignancies. Blood
2013;122(11):1987-9. https://doi.org/10.1182/blood-2013-04-499319 PMid:24030261
- Babushok
DV, Bessler M and Olson T. Genetic predisposition to myelodysplastic
syndrome and acute myeloid leukemia in children and young adults. Leuk
Lymphoma 2016; 57(3):520-36 https://doi.org/10.3109/10428194.2015.1115041 PMid:26693794 PMCid:PMC4798888
[TOP]