Sara Grammatico, Emilia Scalzulli and Maria Teresa Petrucci
Hematology, Department of Cellular Biotechnologies and Hematology, “Sapienza” University, Rome, Italy
Corresponding
author: Maria Teresa Petrucci, MD,
Hematology, Department of Cellular Biotechnologies and Hematology, Via
Benevento 6, 00161 Rome, Italy. Tel +39 06 49974 430, Fax +39 06
441639810. E-mail:
petrucci@bce.uniroma1.it
Published: August 23, 2017
Received: May 16, 2017
Accepted: July 21, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017052 DOI
10.4084/MJHID.2017.052
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Solitary
plasmacytoma is a rare disease characterized by a localized
proliferation of neoplastic monoclonal plasma cells, without evidence
of systemic disease. It can be subdivided into solitary bone
plasmacytoma if the lesion originates in bone, or solitary
extramedullary plasmacytoma if the lesion involves a soft tissue. The
incidence of solitary bone plasmacytoma is higher than solitary
extramedullary plasmacytoma. Also, the prognosis is different: even if
both forms respond well to treatment, overall survival and
progression-free survival of solitary bone plasmacytoma are poorer than
solitary extramedullary plasmacytoma due to its higher rate of
evolution in multiple myeloma. However, the recent advances in the
diagnosis of multiple myeloma can better refine also the diagnosis of
plasmacytoma. Flow cytometry studies and molecular analysis may reveal
clonal plasma cells in the bone marrow; magnetic resonance imaging or
18 Fluorodeoxyglucose positron emission tomography could better define
osteolytic bone lesions. A more explicit exclusion of possible occult
systemic involvement can avoid cases of misdiagnosed multiple myeloma
patients, which were previously considered solitary plasmacytoma and
less treated, with an unavoidable poor prognosis.
Due to the
rarity of the disease, there is no uniform consensus about prognostic
factors and treatment. Radiotherapy is the treatment of choice;
however, some authors debate about the radiotherapy dose and the
relationship with the response rate. Moreover, the role of surgery and
chemotherapy is still under debate. Nevertheless, we must consider that
the majority of studies include a small number of patients and analyze
the efficacy of conventional chemotherapy; few cases are reported
concerning the efficacy of novel agents.
|
Incidence, Signs and Symptoms and Diagnostic Criteria
Solitary
plasmacytoma (SP) is an uncommon type of plasma cell (PC) dyscrasia
that accounts for approximately 2-5% of all PC disorders.[1-3]
It is characterized by a localized proliferation of neoplastic
monoclonal PCs, with no radiologic evidence of additional skeletal
lesions, absence of signs and symptoms of multiple myeloma (MM),
present in the CRAB manifestation (hypercalcemia, renal insufficiency,
anemia and/or bone lesions), and a bone marrow examination
morphologically normal or having a very low clonal PC infiltration
(less than 10%).[4,5]
Due to the rarity of this
disease, there are few studies about it and not a true consensus about
the prognostic factors and treatment. Here, a literature review was
performed to identify relevant articles about SP. PubMed, National
Guideline Clearinghouse Cochrane Database of Systematic Reviews and
ClinicalTrials.gov electronic databases were used for the search. Also
oncology and hematology conference proceedings (European Hematology
Association (EHA), American Society of Clinical Oncology (ASCO),
American Society of Hematology (ASH) and International Myeloma Working
Group (IMW) were used. The search was restricted to documentation
published in human subjects. Case reports were excluded. In Table 1 A and 1 B the key points of the most relevant articles (limited to those published over the last 20 years) are summarized.
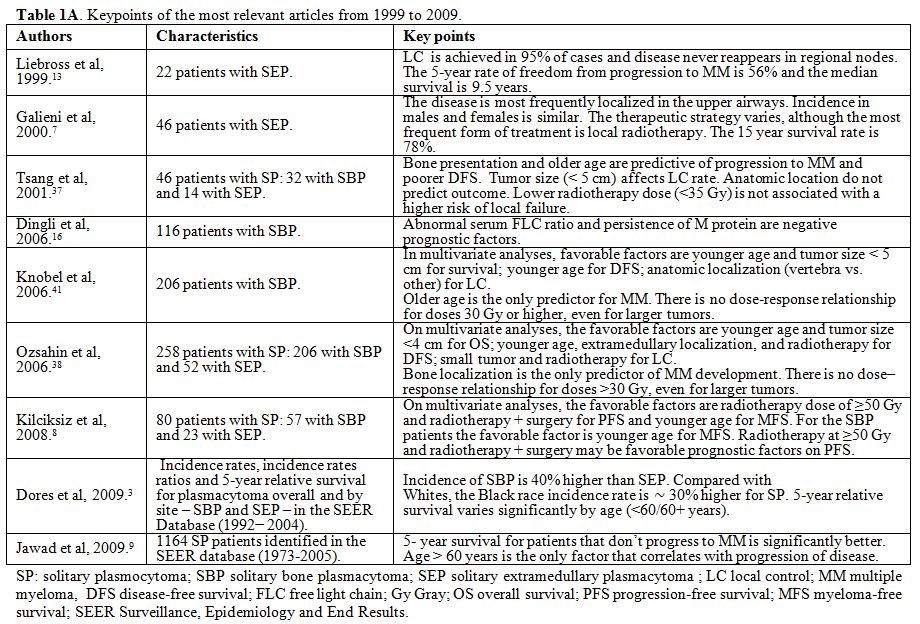 |
Table 1A.
Keypoints of the most relevant articles from 1999 to 2009. |
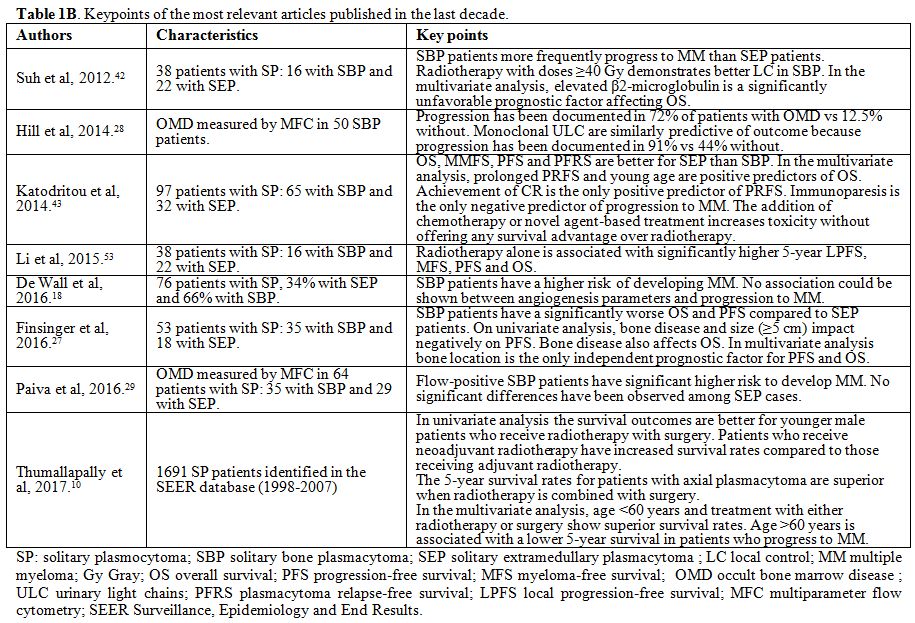 |
Figure 1B. Keypoints of the most relevant articles published in the last decade. |
SP
can be subdivided into two entities: solitary bone plasmacytoma (SBP)
and solitary extramedullary plasmacytoma (SEP), depending on whether
the lesion originates in bones or soft tissues.[6] The incidence of SBP is approximately 40% higher than SEP.[3]
The
median age at diagnosis of SP is 55-60 years, significantly lower than
in MM patients; the male to female ratio varies from 1,2:1 to 2:1.[3,6-10]
Some authors report a higher incidence rate of SP in the black race, around 30% more than white race;[3,9,10] African American to Caucasian incidence rates ratio to develop SP was found to be approximately 1.25/1.30. [9,10] Incidence in Asian population is lower than previous ones; however, its cohort of patients studied is small (about 5.4%).[3] In fact, it is important to consider that the majority of studies refer to a predominantly Caucasian population.
SBP
most frequently occurs in the axial skeleton, such as a vertebra, while
SEP is observed in the head and neck, and these two entities have a
clinical course and prognosis that are quite different from each other.[2,3,5-7,11-17]
All
these data reported are prevalently based on historical studies; there
are few series reported in the most recent years. However, last year,
de Waal et al. reported the experience between 1988 and 2011 in the
northern area of the Netherlands about 76 SP patients, confirming the
historical data. Median age was 61 years (range 26–87), and 60% were
male; 66% had an SBP, localized in the axial skeletal in 78% of these
patients. In the SEP patients, the plasmacytoma was most frequently
located in the oropharynx or nasopharynx (65%).[18]
In
the largest and most recent cohort analyzed, Thumallapally et al.
published a retrospective study of 1691 SP diagnosed in the United
States of America (USA) from 1998 to 2007, analyzing and reporting data
from the Surveillance, Epidemiology and End Results (SEER) database, a
population-based registry in the USA. The median age at diagnosis was
60.38 ± 14.22 years. The male to female ratio was 1.7:1. The cohort was
predominantly Caucasian (80%) followed by African American (14%) and
other races (5.4%). More favorable outcomes were recorded for
Caucasians and patients of other races relative to the African American
cohort, but these differences were not statistically significant (59.1%
vs. 57.6%, p = .083). Bone was the most common site of involvement
(57.78%). Additionally, 831 (49%) and 146 (8%) patients had axial and
appendicular skeletal involvement, respectively, while PC proliferation
in soft tissues was noted in 540 patients (32%). In patients with SEP,
the most commonly encountered site was the upper airway tract (12%).[10]
Signs
and symptoms that patients usually refer are a pain due to: bone
destruction, spinal cord and/or nerve root compression or compression
and enlargement of the soft tissue involved.
Diagnosis of SP needs:
• history and physical examination,
• complete blood count, white blood cell differential, platelet count,
• serum chemistry for creatinine, albumin, corrected calcium,
• serum LDH and Beta-2 microglobulin,
• serum quantitative immunoglobulins and serum protein electrophoresis,
• 24-h urine for total protein, Bence Jones protein, and urine protein electrophoresis,
• serum Free Light Chain (FLC) assay,
• bone marrow aspiration and biopsy,
• skeletal
survey [at least radiography, and/or computed tomography (CT), magnetic
resonance imaging (MRI) or 18 Fluorodeoxyglucose positron emission
tomography (FDG-PET)].
In particular, diagnosis of SBP requires
a solitary bone lesion confirmed by the skeletal survey, the clonal PC
infiltration lower than 10% proven by biopsy, and lack of
myeloma-related organ dysfunction.
Diagnosis of SEP requires a
tissue biopsy indicating monoclonal PC histology, bone marrow clonal PC
infiltration less than 10% of all nucleated cells, the absence of
osteolytic bone lesions or other tissue involvement without CRAB.
Recent
advances have improved the precision of diagnosis. Flow cytometry
studies and molecular detection of heavy- and light-chain gene
rearrangements may reveal clonal PCs in the bone marrow. MRI or FDG-PET
that are more sensitive tests than conventional skeletal survey could
better define patients with systemic disease at diagnosis.[19]
In addition, quantitation of kappa and lambda chains not bound to
intact immunoglobulin molecules in the serum (FLCs), can define more
precisely the diagnosis of MM. This test allows the determination of
clonality based on the involved/uninvolved serum FLCs ratio, which
needs to be ≥100 according to the most recent diagnostic criteria of
MM.[20]
Solitary extramedullary and bone
plasmacytoma show non-specific CT and MRI imaging findings. Usually,
the MRI appearance of SBP is consistent with that of a focal area of
bone marrow replacement; the signal intensity is similar to muscle on
T1-weighted images and hyperintense about muscle on T2-weighted images.
MRI is the modality of choice for soft tissue evaluation; also, MRI of
the axial skeleton has been shown to be superior to whole body X-rays
and is recommended in patients with SBP of the spine and suspected cord
or nerve root compression.[21] FDG-PET is the modality of choice for assessment of the skeletal abnormalities.[22-25]
Recently an abnormal involved serum FLC value and the presence of at
least two hypermetabolic lesions on FDG PET at diagnosis of SP were
reported as the two predictors of early evolution.[26]
Some
patients with SP have a small monoclonal protein (also called M
protein) detectable in the serum and/or in the urine; more frequently
SBP than SEP. The percentage in SBP varies from 24% to 72% of patients
in different reports; levels of uninvolved immunoglobulins are usually
normal.[6,16] In SEP M protein is detected less frequently (about 20% of cases).[7,13,27]
Moreover, Dingli et al. reported in 116 SBP that the FLC ratio was
normal in 53% patients and abnormal in 47%. Patients with an abnormal
FLC had a higher incidence of monoclonal protein in the urine (p <
0.001) and a larger serum M- protein (p = 0.04).[16]
Finally,
Hill e Paiva reported the importance of multiparameter flow cytometry
(MFC) for detecting clonal PCs in the bone marrow;[28,29]
the presence of “minimal occult” bone marrow disease indicates a high
risk of progression for SBP patients and could suggest a tailored of
patients’ follow-up. Conversely, flow diagnostic criteria may also
allow the accurate identification of “true” SP, characterized by
flow-negative bone marrow and absence of M protein, which would
represent a signature for the possibility of cure.
Therefore,
nowadays, a correct diagnosis of SP must include the modern techniques
as MFC and FLC detection in addition to FDG-PET or MRI for the study of
bone lesions. Here we present a possible diagnostic algorithm for SP
divided into three different steps of examinations (Figure 1).
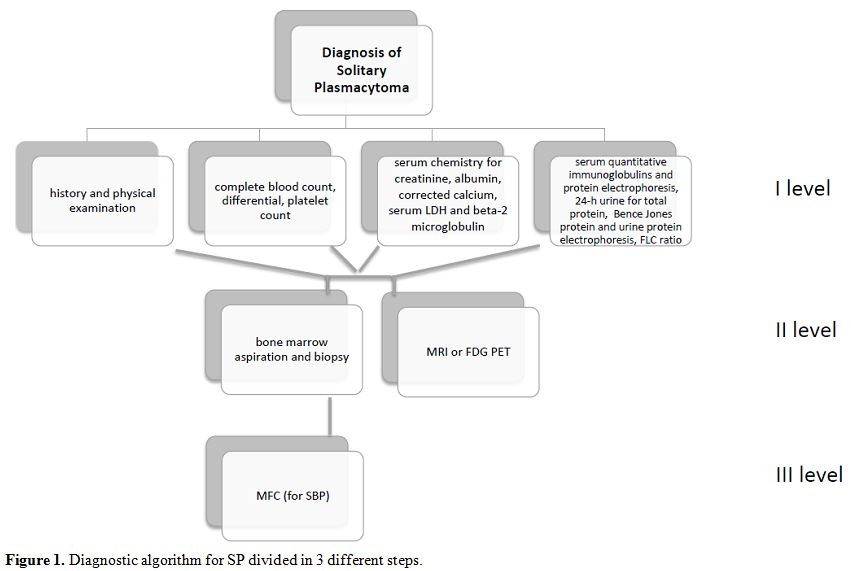 |
Figure 1. Diagnostic algorithm for SP divided in 3 different steps. |
Prognostic Factors
For
SP, it is hard to identify prognostic factors. Several authors reported
various parameters that influence the outcome of these patients, such
as age, lesion size, localization (bone or soft tissue) and the
presence of M protein at the onset and that could disappear after
therapy.[5,13,17,22,30-39] However, these adverse prognostic features have not been consistent between series.[13,31,36,40,41]
In
our previous study, in univariate analysis, the only adverse prognostic
factor for overall survival (OS) was the bone localization rather than
the extramedullary localization. In fact, we could demonstrate a
statistically significant difference between the two groups regarding
5- and 10-year OS probability, with SEP showing a longer survival
probability. Regarding progression-free survival (PFS), the two adverse
prognostic factors in univariate analysis were bone disease and the
larger size of the lesion (>5 cm). In multivariate analysis, the
only significant independent prognostic factor was the bone
localization compared to the extramedullary localization, both
regarding OS and PFS. It must underlie that we identified some
important differences between SBP and SEP patients, particularly a
prevalence of serum M protein and larger tumor size in SBP. We did not
observe significant differences in terms of OS and PFS between patients
with age greater or lower than 60 years.[27]
SBP versus (vs.) SEP.
Other authors reported poor prognosis of SBP in comparison with SEP.
Ozsahin et al. reported in SBP a median time to MM development of 21
months (range 2–135), with a 5-year probability of 45%. The 5-year OS,
disease-free survival (DFS) and local control (LC) rate (considered as
the resolution of the tumor mass and associated symptoms in the
treatment area, with no subsequent evidence of local tumor progression)
was 74%, 50%, and 86%, respectively. SBP had a significantly higher
risk of progression to myeloma at a rate of 65–84% in 10 years and
65–100% in 15 years. In spite of a curative treatment, the median time
to progression to MM was 2 to 3 year.[38] However,
some authors reported that the median OS in patients with SBP was 10
years, and 10% to 20% of patients died of unrelated causes.[6]
Also,
a recent review by Suh et al. confirmed that SBP progressed more
frequently to MM compared with SEP (p = 0.02). In fact, the
Myeloma-free survival (MFS) rate of SEP was 71.2% both at 10-year and
5-, whereas the rates of SBP were at 10-year 36.4% and 0%, at 5,
respectively. The median time of MFS in SBP was 36 months, while SEP
was not attained because of its low progression rate to MM. The median
time of progression to MM following diagnosis was 45 (range, 8–142) and
25 (range, 4–108) months in SEP and SBP, respectively. The 5- and
10-year PFS rates for all patients were 43% and 25%, respectively.
Patients with SBP demonstrated worse PFS rates compared with SEP
patients; however, this difference was not statistically significant (p
= 0.16).[42] Also, the Greek experienced recently
reported a 5 and 10-year OS of 92% and 89% in SEP and 86% and 69% in
SBP, respectively (p = 0.2). The 5- and 10-year MFS probability was 90%
and 70% for patients with SEP vs. 59% and 50% for patients with SBP,
respectively (p = 0.054). Overall, the 5- and 10-year OS probability,
plasmacytoma relapse-free survival (PRFS), PFS and MFS was 84% and 78%,
72% and 58%, 58%, and 43%, and 70% and 59%, respectively. In addition,
in this study, impairment of humoral immunity (immunoparesis), defined
as a suppression of at least one uninvolved immunoglobulin (i.e., for
IgG<700 mg/dL, for IgA <70 mg/dL and IgM<40 mg/dL), was the
only negative predictor of progression to MM.[43]
On
the contrary, less than 30% of patients with SEP develop systemic
progression; some groups reported that the median time to progression
to systemic disease is approximately 2 years.[2,33,44]
Liebross et al. reported an LC rate in 95% of patients with SEP. MM
developed in 32%: the 5-year rate of freedom from progression to MM was
56%, and the median survival was 9.5 years.[13]
Galieni et al. achieved complete remissions in 85% patients, 11% of
partial remissions and 4% of non-response to therapy. Local recurrence
or recurrence at other sites occurred in 7.5% and 10% of patients,
respectively. Moreover, 15% of patients developed MM. The 15-year
survival rate was 78%, and the diffusion-free survival was 83%.[7]
Recently
de Wall et al. confirmed all these data in his cohort of 76 SP
patients. Among SBP patients, 70% progressed to MM with a median time
to progression of 19 months (range 5–131). In SEP patients, 3 (12%)
progressed to MM after 6, 33 and 71 months. The 5-year PFS was
significantly different between SBP and SEP (38% vs. 93%, p = 0.0001).
However, the OS between SBP and SEP was not significantly different (p
= 0.294) with an OS of 70% vs. 81% at 5 years and 64% vs. 77% at 10
years, mainly because 4 SEP patients died within 5 years of diagnosis
due to disorders unrelated to MM. No association with progression to MM
was observed except for the location of the plasmacytoma.[18]
Moreover,
this year, the German group examined the survival of patients with rare
PC and plasmacytoid malignancies (SP, MM, and lymphoplasmacytic
lymphoma) in Germany compared to that in the USA in a period from 1998
to 2012. In Germany, patients with SP had a 5-year relative survival
(RS) of 56.5%, with a lower survival for intraosseous (47.7%) versus
extraosseous (62.0%). Five-year RS estimated for USA patients with SP
was higher at 62.3%, with survival for patients with intraosseous SP at
60.4% and extraosseous SP at 67.8%. Survival trends between 2003 to
2007 and 2008 to 2012 were compared. Five-year age-adjusted RS for
patients with LPL/SP increased in Germany, from 69.2% between 2003 and
2007 to 74.2% in the period 2008 to 2012. In the USA, overall 5-year
age-adjusted RS for these conditions went from 73.3% to 76.8%. Similar
increases were seen for both men and women. However, 5-year RS
increased more for patients with LPL with smaller increases observed
for SP. A better identification of SP might be related to the survival
improvement.[45]
Table 2 A and B
summarize the results of the most relevant series of SBP and SEP cases,
highlighting the better results for SEP regarding LC rate, progression
to MM and OS.
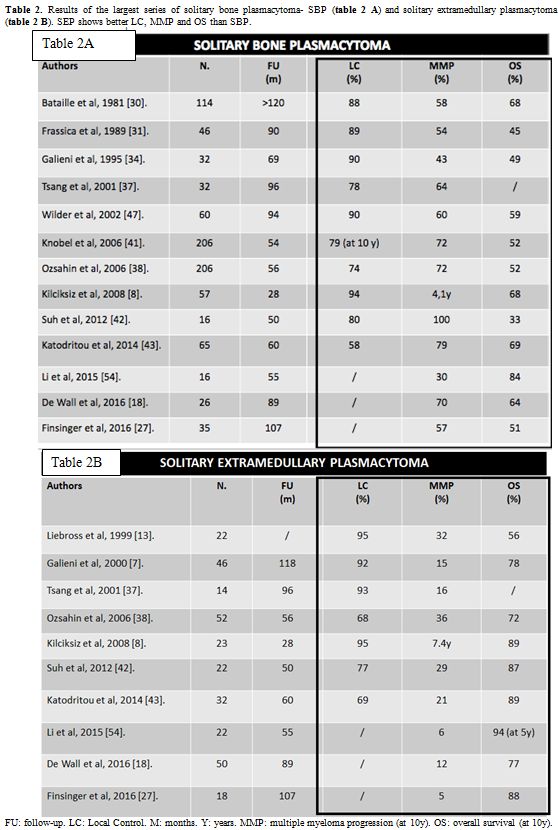 |
Table 2. Results of the largest series of solitary bone plasmacytoma- SBP (table 2 A) and solitary extramedullary plasmacytoma - SEP (table 2 B). SEP shows better LC, MMP and OS than SBP. |
Age, sex, and race.
Numerous studies reported a substantial impact of age on the outcome;
age> than 55 or 60 years is considered poor prognostic factor.[3,9,17,38,41,46]
Recently, Thumallapally et al., in the retrospective analysis on 1691
SP, noted the highest survival rates for the younger age group (<40
years), while patients aged 60 years or older had the most unfavorable
outcomes (87.45% vs. 45%, p < 0.05). Moreover, males had a higher
chance of survival than females (63.7% vs. 52.9%, p < 0.05), and
Caucasians and other races had more favorable outcomes respect to the
African American cohort, but these differences were not statistically
significant (59.1% vs. 57.6%, p = 0.083).[10]
Recently, El-Fattah et al. also reported black race as a poor
prognostic factor.46 However, in the past, Dores did not find
differences in survival between males and females or whites and blacks,[3]
while Jawad reported poorer prognosis for races different from
Caucasian or African American, even if other races represented only the
4.2% of the cohort analyzed in the study.[9]
Tumor size and localization. Different authors agree on the importance of tumor size as a prognostic factor[33,38,41] and the impact of extramedullary involvement vs. bone involvement.[36,38]
For
example, Tsang et al. showed that patients with lesions < 5 cm had
an LC rate of 100%, whereas patients with larger tumors had a rate of
nearly 40%;[37] similar results were obtained by Knobel et al..[41] Other reports suggested a more favorable prognosis for SBP of the appendicular than an axial skeleton in some,[30] but not all studies confirmed this.[41]
In a recent large retrospective USA study, there was no substantial
difference in 5-year relative survival between SBP occurring in the
axial or appendicular skeleton.[3]
M protein and FLC.
Wilder et al. reported that, in multivariate analysis, the persistence
of M protein more than one year after radiotherapy was the only
independent adverse prognostic factor for MFS (p = 0.005) and
cause-specific survival (p = 0.04). Most patients with M protein that
persisted for more than one year after radiotherapy were diagnosed with
MM within 2.2 years of treatment.[30,47]
Dingli
et al. created a risk stratification model based on 2 variables of FLC
ratio and M protein level. Patients with a normal FLC ratio at baseline
and M protein level less than 5 g/L at 1 to 2 years following diagnosis
were considered low risk, patients with either risk factors were
considered intermediate risk and high risk with both risk factors. The
corresponding progression rates at 5 years were significantly different
in the low, intermediate, and high groups: 13%, 26%, and 62%,
respectively (p < 0.001).[16]
Occult Bone Marrow Disease (OMD)
Recent
studies have demonstrated that more careful examination of the bone
marrow in SBP patients identified a clonally related PC population in
68% of the patients. The presence of this occult bone marrow disease
(OMD) is of prognostic significance and is highly predictive of
progression to MM, with a time to progression of 18–26 months.[28,29]
The high predictive value of OMD and the short PFS supports the
potential use of the systemic treatment in addition to local
radiotherapy in this high-risk group of SBP.
Paiva et al. examined
64 patients newly diagnosed with SP, 35 with SBP and 29 with SEP,
respectively. MFC on the bone marrow was performed; an antigen
combination including CD19, CD38, CD45, and CD56 was systematically
evaluated, allowing for identification of the bone marrow PCs
compartment by strong CD38 expression and intermediate side-scatter
signal, and detection of clonal PCs by the recognition of aberrant
phenotypic expression profiles. Patients were defined as flow-positive
when ≥ 20 PCs were detectable by MFC, at a sensitivity level of 10-4.
Clonal PCs were detected in 28 of the 64 patients with SP (44%;
flow-positive), and slightly more frequently in those cases with SBP
(49%) compared with SEP (38%). Among patients with SBP, 71% displaying
clonal PCs progressed into MM, in contrast to only 6% among
flow-negative patients (p < 0.001). Median time to progression (TTP)
was significantly shorter if bone marrow clonality was present (26
months vs. not reached; hazard ratio, 17.4; p < 0.001). Among
patients with SEP, only 20% of flow-positive cases evolved into MM as
compared with 6% of flow-negative patients, and TTP was not
significantly different. Therefore OMD evaluation seems to be relevant
only in SBP cases. It is important to notice that there was no
correlation between the level of M protein and the number of clonal PCs
in the bone marrow.[29]
In the same year, Hill
et al. analyzed 50 patients with SBP with MFC. OMD was defined as a
discrete population of phenotypically aberrant PCs comprising >30%
of total bone marrow PCs. Aberrant phenotype PCs, indicative of OMD,
were demonstrable in 34 of 50 (68%) patients. Progression was
documented in 72% (26/34) of patients with OMD compared with 12.5%
(2/16) without, and the median TTP was 26 months vs not reached (p =
0.003).[28]
The presence of urinary light chains
(ULC) was also predictive of progression because it was documented in
91% (10/11) of patients with ULC vs. 44% (15/34) without (median TTP,
16 vs. 82 months, p < 0.001). When both parameters were assessed,
progression was documented in 75% (24/32) of patients with OMD and/or
ULC but in only 7.7% (1/13) who lacked both parameters (median TTP, 23
months vs. not reached p = 0.001).
However, it is important to have a common flow method for the detection of clonal PCs.[48]
Therapy
Radiotherapy.
Due to the rarity of the disease, there are no randomized studies
regarding the best treatment approach, and the available data from
small case series are somewhat controversial.
Radiotherapy is the treatment of choice for SP, although its efficacy has been tested only in small retrospective series.[5,14,41,49]
So far, no clear relationship has been documented between response and
radiotherapy dose. Some authors suggested a dose of 40-50 Gy for
smaller lesions and >50 Gy for larger lesions (>5cm).[5,14,22,31,41]
In contrast, others authors have shown that <35 Gy are effective for
lesions <5 cm, while lesions >5 cm should be treated with
>40-50 Gy.[32,33,37] The
multicentre study of the Rare Cancer Network, which has analyzed more
than 258 patients with SP and a series of 46 SP patients treated at the
Princess Margaret Hospital, produced no evidence of improvement
regarding LC with radiotherapy doses >30-35 Gy.[37,38]
This datum is in contrast with other series where it is asserted that
radiotherapy doses >45-50 Gy provide better local response rates.[32,33]
In our series, patients received a median dose of 41 Gy (range 21-88),
substantially in agreement with the data reported in the literature
without differences between lower or higher doses of 40 Gy.[27]
Considering
the most recent published works, Kilciksiz et al. reported the 10-year
probability of OS of 73% in all patients with SP, 68% in 57 patients
with SBP, and 89% in 23 patients with SEP, respectively. The
corresponding median PFS values were 3.5 years (95% CI: 2.25–4.81), 3.2
years (95% CI: 1.99–4.37), and 7.4 years (95% CI: 0.57–14.33),
respectively. On multivariate analysis, only the radiation therapy
without surgery and lower radiation doses (<50 Gy) were
significantly associated with lower PFS (p = 0.035 and p = 0.044). The
median MFS values were 4.8 years (%95 CI 1.46–8.24), 4.09 years (%95
CI:2.41–5.77), and 7.45 years (%95 CI: 0.00–14.98) for SP group, SBP
and SEP subgroups, respectively.[17]
At the same
time, Suh et al. didn’t observe a significant dose–response
relationship in patients with SEP; however, radiation doses ≥40 Gy
significantly increased the 5-year LC rates compared to radiation doses
<40 Gy in patients with SBP (100 % vs. 60 %, p = 0.04).[42]
Surgery and radiotherapy.
Even data about the role of the surgery in SP are controversial.
Surgery, a milestone for the histologic diagnosis (biopsy or
partial/total deletion of the lesion), is considered a specific
treatment for plasmacytomas for distinct localizations (spine with
neurological damage, upper airway that cannot be treated with
radiotherapy, or vertebral fractures that require stabilization).[42,50]
Radical
surgery of SEP of the neck and head with curative intent is often a
mutilating procedure; because these tumors are highly radiosensitive,
radical surgery should be avoided. However, for SEP of other sites,
surgical removal, if feasible, could be considered. Because these cases
are rare, it is unclear whether additional treatment with radiotherapy
is necessary for resected SEP with clear surgical margins.[2,17]
Alexiou
et al. reviewed 714 cases with lesion found in the upper aerodigestive
tract and other 155 found in other body regions. Patients with
non-upper aerodigestive tumor had a similar risk of recurrence no
matter they received radiotherapy, surgery or combined treatments.
Interestingly, for upper aerodigestive tract SEP, higher OS and PFS
were detected in those receiving surgery plus radiotherapy.[12]
In addition, a research including 57 SBP and 23 SEP showed that surgery
plus radiotherapy resulted in significantly longer PFS, compared with
radiotherapy alone (median PFS 7.4 years vs. 2.6 years).[8]
Also, Sasaky et al. reported that radiotherapy combined with surgery
was the lone significant prognostic factor for OS of SEP in the head
& neck area (p = 0.04).[50]
More recently,
Gerry et al. reported that SEP in the head & neck area responded
better to surgery than radiotherapy or surgery plus radiotherapy,[51] while some researcher held a contrary opinion.[52,53]
Li
et al. revealed that radiotherapy alone could be considered as a more
effective treatment for SP over surgery; also Jawad et al. did not
report an advantage of surgery alone or radiotherapy plus surgery in
SBP.[54]
Recently, in the retrospective study by
Thumallapally et al. 825 patients (49%) received radiotherapy and 197
(12%) underwent surgery, while 359 patients (21%) required both
radiotherapy and surgery. According to the localization treatment
varied; SBP patients received radiation prevalently only. Patients with
SEP of upper airway tract and the central nervous system received
radiotherapy plus surgery prevalently; whereas those with the lower
airway tract involvement received radiotherapy only or neither
radiotherapy nor surgery, and those with gastrointestinal tract
localization were more frequently treated with surgery only. The
survival rates of patients treated with radiotherapy were significantly
higher than those of patients who did not receive radiotherapy (64.4%
vs. 48.6%, p < 0.05). Moreover, patients who received neoadjuvant
radiotherapy had a greater chance of 5-year relative survival than
those who received adjuvant radiotherapy (86% vs. 73%, p < 0.05). A
significant difference in survival was also observed in patients who
underwent surgery when compared to patients who did not (69.7% vs
57.4%, p < 0.05). Analyzing the 5-year relative survival of
different treatment modalities in relation to the primary site,
patients with axial skeletal involvement treated with a combination of
radiotherapy and surgery had a higher survival rate (70.5%) than those
who received only radiotherapy (61.5%) or surgery (46.4%) (p <
0.05). Patients with upper and lower airway tract involvement had a
higher survival with surgery alone when compared to combination therapy
and radiotherapy (96.7%, 100% p < 0.05 and p < 0.01,
respectively). Patients with involvement of the appendicular skeleton
(63.6%), central nervous system (92.6%) and other sites (excluding
gastrointestinal, skin and connective tissues, lymph nodes) (64.4%) had
an increased survival rate when treated with radiation alone (p =
0.024, p < 0.05, p = 0.004, respectively). In contrast, patients
diagnosed with soft tissue SEP and lymph node SEP obtained better
survival outcomes when received a combination treatment (p = 0.69 and p
= 0.83, respectively), but the differences were not statistically
significant. In the multivariate Cox regression treatment with either
radiotherapy (HR 0.597, p < 0.001) or surgery (HR 0.764 p <
0.001) were all independent predictors of higher OS. Combination
therapy did not appear to be significant in the multivariate analysis
(HR 1.226, 95% CI 0.966-1.552, p = 0.094). Five hundred –fifty-three
patients (32.7%) had progressed to MM during the median follow-up of
9.7 years.[10]
Chemotherapy.
To date, the role of chemotherapy remains controversial; some authors
suggest its combination with local radiotherapy also for patients with
initial SBP. There is only one prospective study that has suggested a
benefit of combined treatment compared to radiotherapy alone.[55]
Here, 53 patients with SBP were randomly assigned to be treated with
either local radiotherapy with doses ranged from 40 to 50 Gy to achieve
LC of disease (28 patients) or the same radiotherapy schedule followed
by melphalan and prednisone given every 6 weeks for 3 years (25
patients). After a median follow-up of 8.9 years, DFS and OS were
improved in patients who were treated with combined therapy; 22
patients remain alive and free of disease in the combined treatment
group compared to only 13 patients in the radiotherapy group (p <
0.01). Another retrospective study showed favorable effects related to
melphalan-based chemotherapy in preventing MM development[32] but in a small series of patients. Other studies did not report any benefits of chemotherapy.[33,35,37,43] Also in our study, we failed to observe significant differences in favor of chemotherapy compared to radiotherapy.[27]
Regarding
the use of novel agents, the Greek group reported the experience of 97
SP. Eighty patients (82.5%) received radiotherapy alone or in
association with chemotherapy or surgery. Twenty-seven patients
received novel agents: particularly 22 bortezomib-based regimens and 5
immunomodulatory drugs (IMiDs). However, the addition of chemotherapy
or novel agents increased toxicity without offering any survival
advantage over radiotherapy.[43]
The Mayo Clinic
group has recently shown a strong correlation between a high degree of
bone marrow angiogenesis and a more rapid progression to MM, in
histological specimens from patients with SBP.[56]
Thus, the use of drugs that inhibit angiogenesis and/or act on the bone
marrow microenvironment (such as IMiDs and proteasome inhibitors) may
represent a novel therapeutic approach also in SP.
Discussion
SBP
and SEP are rare forms of localized PC dyscrasia; these two entities
have a clinical course and prognosis that are quite different from each
other. The updated guidelines recommend to exclude the possibility
of a systemic disease: therefore, all patients with plasmacytoma should
undergo a complete staging that includes: at least whole body X-rays of
the skeleton, bone marrow biopsy and blood tests to rule out full-organ
damage. However, given the recent innovations regarding a diagnosis for
MM, a spine MRI or FDG-PET, serum FLC ratio, immunoglobulin levels and
MFC should be introduced even for the diagnosis of plasmacytoma (Figure 1).
In fact, a better definition of a possible systemic disease, often
underestimated, should reduce the percentage of patients with SP that
evolve in MM. Percentage of evolution in MM is higher in SBP (more than
60%) respect to SEP (less than 30%), with a lower PFS and OS of SBP
than SEP. In general progression to MM develop in 2-3 years.
Due
to the rarity of the disease, there are no randomized studies about the
best treatment approach, and data reported in the literature are
controversial. Radiotherapy is the treatment of choice for SP, but no
clear relationship has been documented between response and
radiotherapy dose. For SBP it is recommended treatment with radical
radiotherapy, with a margin of at least 2 cm and a dose of 40 Gy in 20
fractions. For SBP >5 cm, a higher dose of up to 50 Gy in 25
fractions should be considered.[5]
Surgery is
contraindicated in the absence of structural instability or
neurological compromise. However, due to the development of modern
spinal fixation systems over the last decade, surgical treatment is now
a feasible and successful option for patients who develop pain caused
by structural compromise within the vertebra, vertebral instability,
neurological compromise or both. Loss of structural integrity requires
some forms of stabilization procedure; in cases of neurological
compromise, decompression is also required. The choice of surgery and
approach needs to be tailored to the specific situation of each
patient, the general fitness, and clinical conditions; also the site
and the extent of the tumor must be evaluated. Moreover, if surgery is
required, it should be carried out before starting radiotherapy because
surgery is more difficult in patients who have received radiotherapy
and initial surgery may sometimes compromise radiotherapy, e.g. by the
placing of metal supports.[5]
Patients with a
bulky disease or not responding to radiotherapy could benefit from
chemotherapy; however, studies are limited, and its role is
controversial. The better prognosis of patients with SEP compared to
SBP allows speculating that these are indeed two different diseases
also from a biologic point of view. It is possible to suggest that SEP
patients probably can be treated with surgery or radiotherapy alone,
while SBP patients, who more frequently have an extensive disease,
should be treated with chemotherapy after surgery and/or local
radiotherapy, to prevent disease progression. Moreover, the potential
use of the systemic treatment in addition to local radiotherapy could
be suggested in the high-risk group of SBP with OMD. About
chemotherapy, it is important to consider the possibility of using
novel agents, taking into account the well-documented role that
angiogenesis plays in several hematologic disorders, particularly in PC
dyscrasias. However, there are some small series of cases treated with
bortezomib-based regimens and fewer cases treated with IMiDs.[43,57,58] Here we report a hypothesis for a therapeutic algorithm (Figure 2).
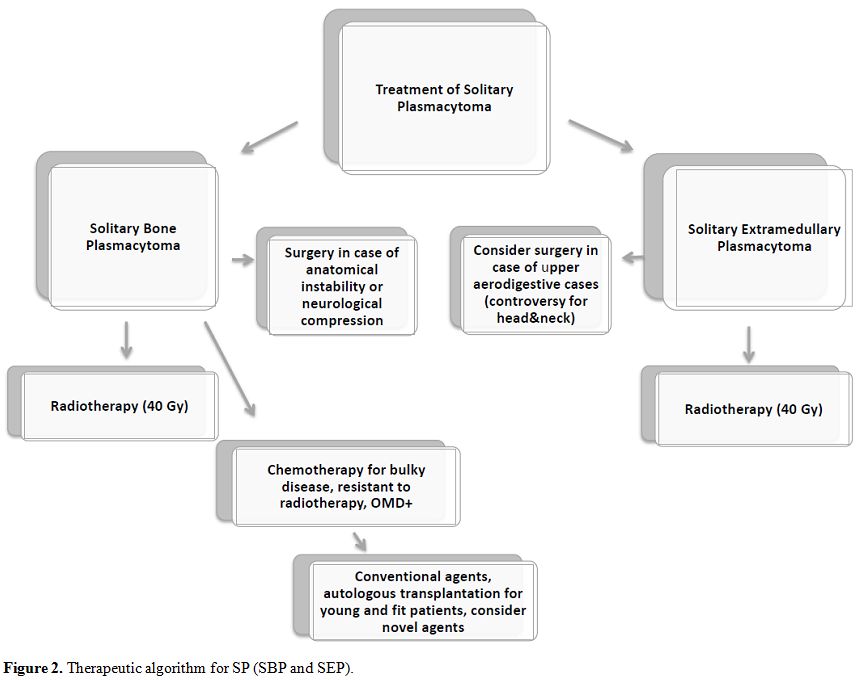 |
Figure 2. Therapeutic algorithm for SP (SBP and SEP). |
Radiotherapy
remains the treatment of choice for both SBP and SEP; surgery should be
indicated only in particular and feasible cases. Chemotherapy should be
considered in cases of bulky disease at the onset or in high-risk
patients.
According to the data reported in the
literature, we can consider high-risk patients cases with abnormal FLC
ratio and M protein level of at least 5 g/L,[16] the presence of OMD,[28,29] or presence of at least two hypermetabolic lesions on FDG-PET.[26]
The patients with disease resistant to radiotherapy could benefit from
chemotherapy too. New drugs could lead to optimal results, as for MM;
also, autologous stem cell transplantation should be considered in
young and fit high-risk or resistant SBP patients. However, larger
prospective clinical studies need to be performed to refine our
understanding of SP (taking into account that SBP and SEP could be
considered as two different entities) and to optimize management of
affected patients, in particular, the role of novel agents for the
treatment of this disease.
References
- Knowling MA, Harwood AR, Bergsagel DE.
Comparison of extramedullary plasmacytomas with solitary and multiple
plasma cell tumors of bone. J Clin Oncol. 1983;1:255-62. https://doi.org/10.1200/JCO.1983.1.4.255 PMid:6668499

- Dimopoulos MA1, Hamilos G. Solitary bone plasmacytoma and extramedullary plasmacytoma. Curr Treat Options Oncol. 2002;3:255-9. https://doi.org/10.1007/s11864-002-0015-2
- Dores
GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS.
Plasmacytoma of bone, extramedullary plasmacytoma, and multiple
myeloma: incidence and survival in the United States, 1992-2004. Br J
Haematol. 2009;144:86-94. https://doi.org/10.1111/j.1365-2141.2008.07421.x PMid:19016727 PMCid:PMC2610331
- Rajkumar
SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S,
Hillengass J, Kastritis E, Richardson P, Landgren O, Paiva B,
Dispenzieri A, Weiss B, LeLeu X, Zweegman S, Lonial S, Rosinol
L, Zamagni E, Jagannath S, Sezer O, Kristinsson SY, Caers J,
Usmani SZ, Lahuerta JJ, Johnsen HE, Beksac M, Cavo M, Goldschmidt H,
Terpos E, Kyle RA, Anderson KC, Durie BG, Miguel JF. International
Myeloma Working Group updated criteria for the diagnosis of multiple
myeloma. Lancet Oncol. 2014;15:e538–48. https://doi.org/10.1016/S1470-2045(14)70442-5
- Soutar
R, Lucraft H, Jackson G, Reece A, Bird J, Low E, Samson D; Guidelines
Working Group of the UK Myeloma Forum.; British Committee for Standards
in Haematology.; British Society for Haematology. Guidelines on the
diagnosis and management of solitary plasmacytoma of bone and solitary
extramedullary plasmacytoma. Br J Haematol. 2004;124:717-26. https://doi.org/10.1111/j.1365-2141.2004.04834.x PMid:15009059
- Dimopoulos
MA, Moulopoulos LA, Maniatis A, Alexanian R. Solitary plasmacytoma of
bone and asymptomatic multiple myeloma. Blood. 2000;96:2037-44.
PMid:10979944
- Galieni
P, Cavo M, Pulsoni A, Avvisati G, Bigazzi C, Neri S, Caliceti U, Benni
M, Ronconi S, Lauria F. Clinical outcome of extramedullary
plasmacytoma. Haematologica. 2000;85:47-51. PMid:10629591
- Kilciksiz
S, Celik OK, Pak Y, Demiral AN, Pehlivan M, Orhan O, Tokatli F, Agaoglu
F, Zincircioglu B, Atasoy BM, Ozseker N, Yersal O, Niang U, Haydaroglu
A; Turkish Oncology Group-Sarcoma Working Party.Clinical and prognostic
features of plasmacytomas: a multicenter study of Turkish Oncology
Group-Sarcoma Working Party. Am J Hematol. 2008;83:702-7. https://doi.org/10.1002/ajh.21211 PMid:18543343
- Jawad
MU, Scully SP. Skeletal Plasmacytoma: progression of disease and impact
of local treatment; an analysis of SEER database. J Hematol Oncol.
2009;2:41. https://doi.org/10.1186/1756-8722-2-41 PMid:19778427 PMCid:PMC2759950
- Thumallapally
N, Meshref A, Mousa M, Terjanian T. Solitary plasmacytoma:
population-based analysis of survival trends and effect of various
treatment modalities in the USA. BMC Cancer. 2017;17:13. https://doi.org/10.1186/s12885-016-3015-5 PMid:28056880 PMCid:PMC5216567
- Wiltshaw
E. The natural history of extramedullary plasmacytoma and its relation
to solitary myeloma of bone and myelomatosis. Medicine (Baltimore).
1976;55:217-38. https://doi.org/10.1097/00005792-197605000-00002
- Alexiou
C, Kau RJ, Dietzfelbinger H, Kremer M, Spiess JC, Schratzenstaller B,
Arnold W. Extramedullary plasmacytoma: tumor occurrence and therapeutic
concepts. Cancer. 1999;85:2305-14. https://doi.org/10.1002/(SICI)1097-0142(19990601)85:11<2305::AID-CNCR2>3.0.CO;2-3
- Liebross
RH, Ha CS, Cox JD, Weber D, Delasalle K, Alexanian R. Clinical course
of solitary extramedullary plasmacytoma. Radiother Oncol.
1999;52:245-9. https://doi.org/10.1016/S0167-8140(99)00114-0
- Hu
K1, Yahalom J. Radiotherapy in the management of plasma cell tumors.
Oncology (Williston Park). 2000;14:101-8, 111; discussion 111-2, 115.
- Chao
MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the
management of solitary extramedullary plasmacytoma. Intern Med J. 2005
Apr;35(4):211-5. https://doi.org/10.1111/j.1445-5994.2005.00804.x PMid:15836498
- Dingli
D, Kyle RA, Rajkumar SV, Nowakowski GS, Larson DR, Bida JP, Gertz MA,
Therneau TM, Melton LJ 3rd, Dispenzieri A, Katzmann JA. Immunoglobulin
free light chains and solitary plasmacytoma of bone. Blood.
2006;108:1979-83. https://doi.org/10.1182/blood-2006-04-015784 PMid:16741249 PMCid:PMC1895544
- Kilciksiz
S, Karakoyun-Celik O, Agaoglu FY, Haydaroglu A. A review for solitary
plasmacytoma of bone and extramedullary plasmacytoma. Scientific World
Journal. 2012;2012:895765. doi: 10.1100/2012/895765. https://doi.org/10.1100/2012/895765
- de
Waal EG, Leene M, Veeger N, Vos HJ, Ong F, Smit WG, Hovenga S,
Hoogendoorn M, Hogenes M, Beijert M, Diepstra A, Vellenga E.
Progression of a solitary plasmacytoma to multiple myeloma. A
population-based registry of the northern Netherlands. Br J Haematol.
2016;175:661-7. https://doi.org/10.1111/bjh.14291 PMid:27605358
- Moulopoulos
LA, Dimopoulos MA, Weber D, Fuller L, Libshitz HI, Alexanian R.
Magnetic resonance imaging in the staging of solitary plasmacytoma of
bone. J Clin Oncol. 1993;11:1311-5. https://doi.org/10.1200/JCO.1993.11.7.1311 PMid:8315427
- Dispenzieri
A, Kyle R, Merlini G, Miguel JS, Ludwig H, Hajek R, Palumbo A,
Jagannath S, Blade J, Lonial S, Dimopoulos M, Comenzo R, Einsele H,
Barlogie B, Anderson K, Gertz M, Harousseau JL, Attal M, Tosi P,
Sonneveld P, Boccadoro M, Morgan G, Richardson P, Sezer O, Mateos MV,
Cavo M, Joshua D, Turesson I, Chen W, Shimizu K, Powles R, Rajkumar SV,
Durie BG; International Myeloma Working Group. International Myeloma
Working Group guidelines for serum-free light chain analysis in
multiple myeloma and related disorders. Leukemia, 2009; 23:215-224. https://doi.org/10.1038/leu.2008.307 PMid:19020545
- Dimopoulos
MA, Hillengass J, Usmani S, Zamagni E, Lentzsch S, Davies FE, Raje N,
Sezer O, Zweegman S, Shah J, Badros A, Shimizu K, Moreau P, Chim CS,
Lahuerta JJ, Hou J, Jurczyszyn A, Goldschmidt H, Sonneveld P, Palumbo
A, Ludwig H, Cavo M, Barlogie B, Anderson K, Roodman GD, Rajkumar SV,
Durie BG, Terpos E. Role of magnetic resonance imaging in the
management of patients with multiple myeloma: a consensus statement. J
Clin Oncol 2015;33: 657–64. https://doi.org/10.1200/JCO.2014.57.9961 PMid:25605835
- Vogl
TJ1, Steger W, Grevers G, Balzer J, Mack M, Felix R. MR characteristics
of primary extramedullary plasmacytoma in the head and neck. AJNR Am J
Neuroradiol. 1996 Aug;17(7):1349-54. PMid:8871723
- Lu
YY, Chen JH, Lin WY, Liang JA, Wang HY, Tsai SC, Kao CH. FDG PET or
PET/CT for detecting intramedullary and extramedullary lesions in
multiple Myeloma: a systematic review and meta-analysis. Clin Nucl Med
2012;37:833–37. https://doi.org/10.1097/RLU.0b013e31825b2071 PMid:22889770
- Agarwal A. Neuroimaging of plasmacytoma. A pictorial review. Neuroradiol J. 2014;27:431-7. https://doi.org/10.15274/NRJ-2014-10078 PMid:25196616 PMCid:PMC4236876
- Cavo
M, Terpos E, Nanni C, Moreau P, Lentzsch S, Zweegman S, Hillengass J,
Engelhardt M, Usmani SZ, Vesole DH, San-Miguel J, Kumar SK, Richardson
PG, Mikhael JR, da Costa FL, Dimopoulos MA, Zingaretti C, Abildgaard N,
Goldschmidt H, Orlowski RZ, Chng WJ, Einsele H, Lonial S, Barlogie B,
Anderson KC, Rajkumar SV, Durie BG, Zamagni E. Role of 18F-FDG PET/CT
in the diagnosis and management of multiple myeloma and other plasma
cell disorders: a consensus statement by the International Myeloma
Working Group. Lancet Oncol. 2017;18:e206-e217. doi:
10.1016/S1470-2045(17)30189-4. https://doi.org/10.1016/S1470-2045(17)30189-4
- Fouquet
G, Guidez S, Herbaux C, Van de Wyngaert Z, Bonnet S, Beauvais D,
Demarquette H, Adib S, Hivert B, Wemeau M, Berthon C, Terriou L,
Coiteux V, Macro M, Decaux O, Facon T, Huglo D, Leleu X. Impact of
initial FDG-PET/CT and serum-free light chain on transformation of
conventionally defined solitary plasmacytoma to multiple myeloma. Clin
Cancer Res. 2014;20:3254-60. https://doi.org/10.1158/1078-0432.CCR-13-2910 PMid:24714772
- Finsinger
P, Grammatico S, Chisini M, Piciocchi A, Foà R, Petrucci MT. Clinical
features and prognostic factors in solitary plasmacytoma. Br J
Haematol. 2016;172:554-60. https://doi.org/10.1111/bjh.13870 PMid:26684545
- Hill
QA, Rawstron AC, de Tute RM, Owen RG. Outcome prediction in
plasmacytoma of bone: a risk model utilizing bone marrow flow cytometry
and light-chain analysis. Blood. 2014;124:1296-9. https://doi.org/10.1182/blood-2014-04-566521 PMid:24939658
- Paiva
B, Chandia M, Vidriales MB, Colado E, Caballero-Velázquez T, Escalante
F, Garcia de Coca A, Montes MC, Garcia-Sanz R, Ocio EM, Mateos MV, San
Miguel JF. Multiparameter flow cytometry for staging of solitary bone
plasmacytoma: new criteria for risk of progression to myeloma. Blood.
2014;124:1300-3. https://doi.org/10.1182/blood-2014-04-567909 PMid:24876564
- Bataille
R, Sany J, Serre H. Apparently isolated plasmacytoma of bone. Clinical
and prognostic data. 114 cases and review of literature (author's
transl). Nouv Presse Med. 1981;10:407-11. PMid:7220335
- Frassica
DA, Frassica FJ, Schray MF, Sim FH, Kyle RA. Solitary plasmacytoma of
bone: Mayo Clinic experience. Int J Radiat Oncol Biol Phys.
1989;16:43-8. https://doi.org/10.1016/0360-3016(89)90008-4
- Mayr
NA, Wen BC, Hussey DH, Burns CP, Staples JJ, Doornbos JF, Vigliotti AP.
The role of radiation therapy in the treatment of solitary
plasmacytomas. Radiother Oncol. 1990;17:293-303. https://doi.org/10.1016/0167-8140(90)90003-F
- Holland
J, Trenkner DA, Wasserman TH, Fineberg B. Plasmacytoma. Treatment
results and conversion to myeloma. Cancer. 1992;69:1513-7. https://doi.org/10.1002/1097-0142(19920315)69:6<1513::AID-CNCR2820690633>3.0.CO;2-X
- Galieni
P, Cavo M, Avvisati G, Pulsoni A, Falbo R, Bonelli MA, Russo D,
Petrucci MT, Bucalossi A, Tura S. Solitary plasmacytoma of bone and
extramedullary plasmacytoma: two different entities? Ann Oncol.
1995;6:687-91. https://doi.org/10.1093/oxfordjournals.annonc.a059285 PMid:8664190
- Shih
LY, Dunn P, Leung WM, Chen WJ, Wang PN. Localised plasmacytomas in
Taiwan: comparison between extramedullary plasmacytoma and solitary
plasmacytoma of bone. Br J Cancer. 1995;71:128-33. https://doi.org/10.1038/bjc.1995.26 PMid:7819027 PMCid:PMC2033434
- Bolek
TW1, Marcus RB Jr, Mendenhall NP. Solitary plasmacytoma of bone and
soft tissue. Int J Radiat Oncol Biol Phys. 1996;36:329-33. https://doi.org/10.1016/S0360-3016(96)00334-3
- Tsang
RW, Gospodarowicz MK, Pintilie M, Bezjak A, Wells W, Hodgson DC,
Stewart AK. Solitary plasmacytoma treated with radiotherapy: impact of
tumor size on outcome. Int J Radiat Oncol Biol Phys. 2001;50:113-20. https://doi.org/10.1016/S0360-3016(00)01572-8
- Ozsahin
M, Tsang RW, Poortmans P, Belkacémi Y, Bolla M, Dinçbas FO, Landmann C,
Castelain B, Buijsen J, Curschmann J, Kadish SP, Kowalczyk A, Anacak Y,
Hammer J, Nguyen TD, Studer G, Cooper R, Sengöz M, Scandolaro L,
Zouhair A. Outcomes and patterns of failure in solitary plasmacytoma: a
multicenter Rare Cancer Network study of 258 patients. Int J Radiat
Oncol Biol Phys. 2006;64:210-7. https://doi.org/10.1016/j.ijrobp.2005.06.039 PMid:16229966
- Dagan R1, Morris CG, Kirwan J, Mendenhall WM. Solitary plasmacytoma. Am J Clin Oncol. 2009;32:612-7. https://doi.org/10.1097/COC.0b013e31819cca18 PMid:19593082
- Chak
LY, Cox RS, Bostwick DG, Hoppe RT. Solitary plasmacytoma of bone:
treatment, progression, and survival. J Clin Oncol. 1987;5:1811-5. https://doi.org/10.1200/JCO.1987.5.11.1811 PMid:3681369
- Knobel
D, Zouhair A, Tsang RW, Poortmans P, Belkacémi Y, Bolla M, Oner FD,
Landmann C, Castelain B, Ozsahin M; Rare Cancer Network. Prognostic
factors in solitary plasmacytoma of the bone: a multicenter Rare Cancer
Network study. BMC Cancer. 2006;6:118. https://doi.org/10.1186/1471-2407-6-118 PMid:16677383 PMCid:PMC1479355
- Suh
YG, Suh CO, Kim JS, Kim SJ, Pyun HO, Cho J. Radiotherapy for solitary
plasmacytoma of bone and soft tissue: outcomes and prognostic factors.
Ann Hematol. 2012;91:1785-93. https://doi.org/10.1007/s00277-012-1510-6 PMid:22752147
- Katodritou
E, Terpos E, Symeonidis AS, Pouli A, Kelaidi C, Kyrtsonis MC,
Kotsopoulou M, Delimpasi S, Christoforidou A, Giannakoulas N, Viniou
NA, Stefanoudaki E, Hadjiaggelidou C, Christoulas D, Verrou E, Gastari
V, Papadaki S, Polychronidou G, Papadopoulou A, Giannopoulou E,
Kastritis E, Kouraklis A, Konstantinidou P, Anagnostopoulos A, Zervas
K, Dimopoulos MA. Clinical features, outcome, and prognostic factors
for survival and evolution to multiple myeloma of solitary
plasmacytomas: a report of the Greek myeloma study group in 97
patients. Am J Hematol. 2014;89:803-8. https://doi.org/10.1002/ajh.23745 PMid:24757085
- Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005:373-6. https://doi.org/10.1182/asheducation-2005.1.373 PMid:16304406
- Weberpals
J, Pulte D, Jansen L, Luttmann S, Holleczek B, Nennecke A, Ressing M,
Katalinic A, Merz M, Brenner H for the GEKID Cancer Survival Working
Group. Survival of patients with lymphoplasmacytic lymphoma and
solitary plasmacytoma in Germany and the United States of America in
the early 21st century. Haematologica 2017; 102:e229
- El-Fattah
MA, Aboelmagd M, Elhamouly M. Clinical risk factors of Plasmacytoma
mortality: a US population-based study. Br J Haematol. 2016 Jun 13.
doi: 10.1111/bjh.14189. https://doi.org/10.1111/bjh.14189
- Wilder
RB, Ha CS, Cox JD, Weber D, Delasalle K, Alexanian R. Persistence of
myeloma protein for more than one year after radiotherapy is an adverse
prognostic factor in solitary plasmacytoma of bone. Cancer.
2002;94:1532-1537. https://doi.org/10.1002/cncr.10366 PMid:11920511
- Dimopoulos MA, Terpos E. Solitary bone plasmacytomas need to flow. Blood. 2014;124:1209-10. https://doi.org/10.1182/blood-2014-06-579706 PMid:25147376 PMCid:PMC4141506
- Mendenhall WM, Mendenhall CM, Mendenhall NP. Solitary plasmacytoma of bone and soft tissues. Am J Otolaryngol. 2003;24:395-9. https://doi.org/10.1016/S0196-0709(03)00092-9
- Sasaki
R, Yasuda K, Abe E, Uchida N, Kawashima M, Uno T, Fujiwara M, Shioyama
Y, Kagami Y, Shibamoto Y, Nakata K, Takada Y, Kawabe T, Uehara K, Nibu
K, Yamada S. Multi-institutional analysis of solitary extramedullary
plasmacytoma of the head and neck treated with curative radiotherapy.
Int J Radiat Oncol Biol Phys. 2012;82:626-34. https://doi.org/10.1016/j.ijrobp.2010.11.037 PMid:21277117
- Gerry
D, Lentsch EJ. Epidemiologic evidence of superior outcomes for
extramedullary plasmacytoma of the head and neck. Otolaryngol Head Neck
Surg. 2013;148:974-81. https://doi.org/10.1177/0194599813481334 PMid:23482476
- Michalaki
VJ, Hall J, Henk JM, Nutting CM, Harrington KJ. Definitive radiotherapy
for extramedullary plasmacytomas of the head and neck. Br J Radiol.
2003;76:738-41. https://doi.org/10.1259/bjr/54563070 PMid:14512335
- Creach
KM, Foote RL, Neben-Wittich MA, Kyle RA. Radiotherapy for
extramedullary plasmacytoma of the head and neck. Int J Radiat Oncol
Biol Phys. 2009;73:789-94. https://doi.org/10.1016/j.ijrobp.2008.04.077 PMid:18707826
- Li
QW, Niu SQ, Wang HY, Wen G, Li YY, Xia YF, Zhang YJ. Radiotherapy Alone
is Associated with Improved Outcomes Over Surgery in the Management of
Solitary Plasmacytoma. Asian Pac J Cancer Prev. 2015;16:3741-5. https://doi.org/10.7314/APJCP.2015.16.9.3741
- Aviles
A, Huerta-Guzman J, Delgado S, Fernadez A, Diaz-Maqueo JC. Improved
outcome in solitary bone plasmacytoma with combined therapy. Hematol
Oncol 1996, 14:111-117. https://doi.org/10.1002/(SICI)1099-1069(199609)14:3<111::AID-HON575>3.0.CO;2-G
- Kumar
SK, Callander NS, Alsina M, Atanackovic D, Biermann JS, Chandler JC,
Costello C, Faiman M, Fung HC, Gasparetto C, Godby K, Hofmeister C,
Holmberg L, Holstein S, Huff CA, Kassim A, Liedtke M, Martin T, Omel J,
Raje N, Reu FJ, Singhal S, Somlo G, Stockerl-Goldstein K, Treon SP,
Weber D, Yahalom J, Shead DA, Kumar R. Multiple Myeloma, Version
3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr
Canc Netw. 2017;15:230-69. https://doi.org/10.6004/jnccn.2017.0023 PMid:28188192
- Fukuhara
S, Tazawa H, Okanobu H, Kida M, Kido M, Takafuta T, Nishida T, Ohdan H,
Sakimoto H. Successful treatment of primary advanced gastric
plasmacytoma using a combination of surgical resection and chemotherapy
with bortezomib: A case report. Int J Surg Case Rep. 2016;27:133-136. https://doi.org/10.1016/j.ijscr.2016.08.041 PMid:27611798 PMCid:PMC5018075
- Khaliq
W1, Uzoaru I, Konchanin RP, Sapiente RA, Egner JR. Solitary
extramedullary plasmacytoma of the bladder: a case report and
literature. Oncology. 2010;24:832-5. PMid:20923037
[TOP]