Kensuke Matsuda1, Yosuke Matsumoto2, Mihoko Yoshida2, Kazuho Shimura2, Hiroto Kaneko2, Tohru Inaba3, Shigeo Horiike4, Junya Kuroda4 and Masafumi Taniwaki2,5
1 Department of Hematology and Oncology, Tokyo University Hospital, Japan.
2 Departments of Hematology and Laboratory Medicine, Aiseikai Yamashina Hospital, Japan.
3
Department of Infection Control and Laboratory Medicine, Kyoto
Prefectural University of Medicine, Graduate School of Medical Science,
Japan.
4 Division of Hematology and Oncology, Kyoto Prefectural University of Medicine, Graduate School of Medical Science, Japan.
5
Center for Molecular Diagnostics and Therapeutics, Kyoto Prefectural
University of Medicine, Graduate School of Medical Science, Japan.
Corresponding
author: Yosuke Matsumoto, MD, Ph.D.,
Departments of Hematology and Laboratory Medicine, Aiseikai Yamashina
Hospital, 19-4 Takehana-Shichouno-cho, Yamashina-ku, Kyoto 607-8086,
Japan. Fax: +81-75-593-3179; E-mail:
yosuke-m@koto.kpu-m.ac.jp
Published: September 1, 2017
Received: July 26, 2017
Accepted: August 8, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017054 DOI
10.4084/MJHID.2017.054
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Hairy
B-cell lymphoproliferative disorder (HBLD) is one of chronic polyclonal
B-cell lymphocytosis. We report a 47-year-old female Japanese patient
diagnosed as having HBLD based on lymphocytosis with hairy cell
appearance and characteristic phenotypes including CD11c+ and without
B-cell monoclonality. She was a non-smoker and possessed HLA-DR4. She
has been closely followed up without treatment and lymphoma development
for over five years. Although this disease is quite rare and has been
reported, to our knowledge, in only 13 Japanese cases, an accurate
diagnosis, particularly differential diagnosis from persistent
polyclonal B-cell lymphocytosis or hairy cell leukemia-Japanese variant
is essential for the prevention of unnecessary treatments.
|
Introduction
Primary
lymphocytosis is defined as a set of conditions associated with an
increase in the absolute number of lymphocytes secondary to an
intrinsic defect in the expanded lymphocyte population.[1]
These conditions include monoclonal lymphocytic malignancies and
polyclonal B-cell lymphocytosis. A representative form of the latter is
persistent polyclonal B-cell lymphocytosis (PPBL), with more than 100
cases having been reported in western countries.[2-6]
Another form of polyclonal B-cell lymphocytosis is hairy B-cell
lymphoproliferative disorder (HBLD), which is extremely rare with, to
the best of our knowledge, only 13 cases reported, all of them in
Japanese people.[7-14]
For a right diagnosis of
HBLD, distinguishing this disease from hairy cell leukemia-Japanese
variant (HCL-Jv), an accurate workup is needed because of their
morphological similarities including hairy cell appearance and a
characteristic immunophenotype.[8] The essential difference is whether B-cell monoclonality is detected or not.
This
report concerns a female patient with HBLD. The absence of B-cell
clonalities was confirmed regarding the lack of light chain restriction
verified both using flow cytometric analysis and clonal immunoglobulin
(Ig) gene rearrangement determined using Southern blotting analysis and
multiplex polymerase chain reaction (PCR).
Clinical Presentation
A
47-year-old non-smoking Japanese woman visited the University Hospital
Kyoto Prefectural University of Medicine in June 2012 because of
blurred vision, exertional dyspnea, and an uncomfortable feeling in the
throat. Her past medical history was not remarkable. Physical
examination and positron emission tomography/computed tomography
detected hepatomegaly (2 cm below the right costal margin) and
splenomegaly (4 cm below the left costal margin), but no definite signs
of lymphadenopathy. Small retinal bilateral hemorrhages were detected.
Hematological examination showed 10.5 g/dl Hb, 175 x 109 /l platelets, and 23.0 x 109
/l WBC with 67.0% atypical lymphocytes. Peripheral blood smears and
electron microscopic examination found that these atypical lymphocytes
had irregularly shaped abundant cytoplasms with partially hairy
projections (Figure 1).
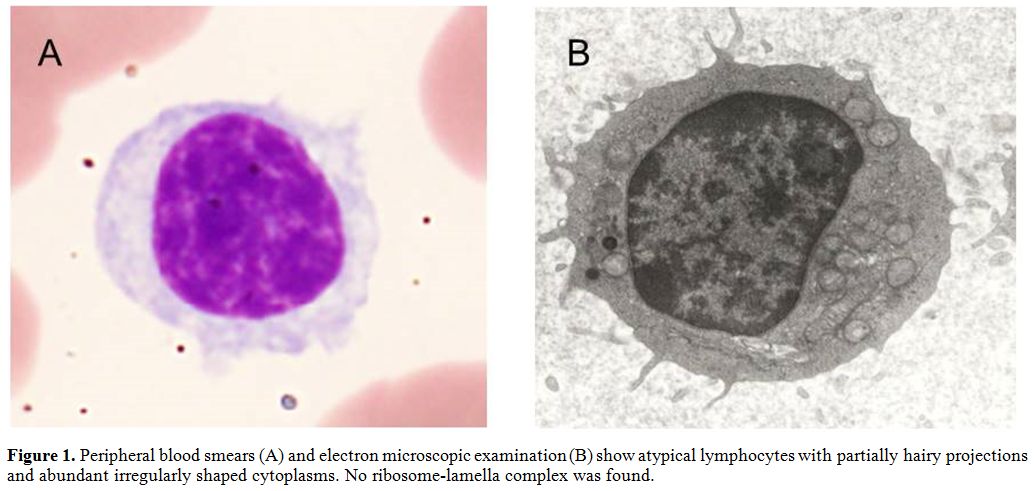 |
Figure 1. Peripheral blood smears (A) and
electron microscopic examination (B) show atypical lymphocytes with
partially hairy projections and abundant irregularly shaped cytoplasms.
No ribosome-lamella complex was found. |
No
ribosome-lamella complex (RLC) was found, but rouleau formation was
detected. The serum level of IgG was 6278 mg/dl, of IgA 359 mg/dl, and
of IgM 283 mg/dl. Fractionation of serum protein showed no M-peak, and
urine and serum immunoelectrophoretic studies showed no M-protein. Flow
cytometric (FCM) analysis of the peripheral blood immunophenotype
showed positivity for CD11c (38.9%), CD19 (64.3%), CD20 (62.5%), and
CD22 (50.5%), and negativity for CD5 (16.6%), CD10 (0.9%), CD23 (0.1%),
CD25 (1.0%), FMC-7 (3.7%), and ZAP-70 (4.4%). No light chain
restriction was detected (k:lambda=33.9%:13.6%).
Southern blotting analysis of peripheral blood cells showed no
rearrangement band for either the clonal immunoglobulin heavy chain
(IGH) gene (JH, Cµ) or the light chain gene (Jk, Ck, Clambda).
Multiplex PCR analysis using the three sets of VH primers and one JH
consensus primer as specified for the BIOMED-2 primer sets[15] showed no monoclonal peak of IGH gene recombination (Figure 2).
Cytogenetic analysis of bone marrow cells using G-banding revealed 46,
XX [11/11]. Direct DNA sequencing showed no BRAF V600E mutation.[16]
Our patient possessed the HLA-DR4/DR5 histocompatibility complex. She
was diagnosed as having HBLD based on the finding of a polyclonal
proliferation of hairy B-cells with phenotypes such as CD5-, CD11c+,
CD20+, and CD25-. Although she has been closely followed up without
treatment, her WBC count has been stable (8.9 x 109/l) (atypical lymphocytes 31.0%), and her general status has remained fair over five years.
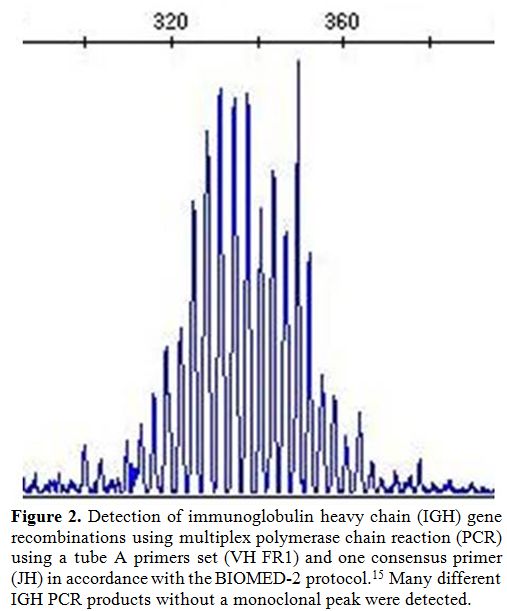 |
Figure 2. Detection of immunoglobulin
heavy chain (IGH) gene recombinations using multiplex polymerase chain
reaction (PCR) using a tube A primers set (VH FR1) and one consensus
primer (JH) in accordance with the BIOMED-2 protocol.[15] Many
different IGH PCR products without a monoclonal peak were detected. |
Discussion
The
subject of this report is a 47-year-old female Japanese patient with
HBLD, which is a very rare disorder so that an accurate diagnosis is
important but difficult. To establish the diagnosis of this case, we
had to exclude PPBL and HCL-Jv. A comparison between the features of
our case, HBLD reported cases, PPBL and HCL-Jv is shown in Table 1.
 |
Table 1. Comparison of previously reported
cases of hairy B-cell lymphoproliferative disorder (HBLD), persistent
polyclonal B-cell lymphocytosis (PPBL), and hairy cell
leukemia-Japanese variant (HCL-Jv). |
According to a
review of previous reports concerning 13 cases of HBLD, the median age
for onset of HBLD was 57 years (range 29-80 years) and the male: female
ratio was 3:10. These cases generally possessed HLA-DR4.[8,12,14] Cases of PPBL, on the other hand, usually are also young to middle-aged women, often smokers and HLA-DR7 positive.[5,6] HBLD and HCL-Jv17,[18] are more prevalent in the elderly than PPBL.[6]
As
to the clinical features of our case, they showed splenomegaly but no
definite signs of lymphadenopathy as mentioned in previous reports of
HBLD,[8-11,13,14] PPBL[6] and HCL-Jv.[17,18]
Laboratory findings of the peripheral blood showed lymphocytosis and
elevated serum IgG level without M-protein. The retinal hemorrhages of
our case were thought to be due to hyperviscosity associated with her
hypergammaglobulinemia. All the reported HBLD cases[7-14] and some of the HCL-Jv cases[18]
had elevated serum IgG levels with a polyclonal pattern but without
M-protein. On the other hand, PPBL cases reportedly had elevated serum
IgM levels.[6] Although this IgM elevation is mostly
polyclonal, Cornet et al. reported that 2 of 111 cases had IgM
monoclonal gammopathy of undetermined significance at the time of PPBL
diagnosis.[6]
Morphologically, the atypical
lymphocytes of our case had a hairy cell appearance and a round
nucleus, but HBLD cases, including our case, did not have RLC. Although
RLC is a specific electron microscopic finding of hairy cell leukemia,
it had been found in only 4 of 26 (15%) HCL-Jv cases.[17]
The immunophenotype of our case was CD5-, CD11c+, CD20+, and CD25-,
which corresponds to previously reported findings of HBLD as well as of
HCL-Jv.[8,18] For these reasons, it
might be impossible to distinguish HBLD from HCL-Jv regarding
morphology and immunophenotype. Moreover, among the PPBL features the
atypical lymphocytes are morphologically binucleated and not villous,
and immunophenotypically CD11c-.[3]
For these
reasons, the clonality analyses are essential for distinguishing HBLD
from HCL-Jv and morphology and immunophenotype from PPBL. No B-cell
clonalities were detected in our case by FCM analysis for light chains,
by Southern blotting and multiplex PCR for the rearrangements of Ig
genes, by cytogenetic analysis using G-banding, and by
immunoelectrophoretic studies for M-protein. Although proliferated
B-cells of both HBLD and PPBL cases are polyclonal, some of the latter
may have small numbers of clonal B-cells. In fact, PPBL can be
associated with recurrent chromosomal abnormalities such as +i(3q)[4,6] or t(14;18)[19] etc. and clonal Ig gene rearrangements.[3,6,20] Furthermore, while there have been no reports on the clonal evolution of HBLD, cases of PPBL can develop B-cell lymphoma.[5,6]
In
conclusion, we reached the diagnosis of HBLD for our case on the basis
of hairy cell appearance, the presence of a characteristic
immunophenotype (CD5-, CD11c+, CD20+, and CD25-), and the absence of
B-cell monoclonality. All previously reported cases of HBLD also
satisfied these features. These findings are useful for the correct
diagnosis of HBLD and for differentiating this disease from PPBL and
HCL-Jv. Of the 14 reported HBLD cases including our case, 7 cases were
from Osaka University,[8,9] and 2 from our Institute.[14]
Until
now and without therapy, our case has not shown any B-cell clonality
nor any malignant development of HBLD. A wait-and-see approach without
treatment can, therefore, be considered a potentially useful strategy
for HBLD. Our case demonstrates that accurate diagnosis of this disease
is essential for the prevention of unnecessary treatments.
References
- Beutler E, Lichtman MA, Coller BS, Kipps TJ,
Seligsohn U, editors. Williams Hematology, sixth ed. New York: McGraw
Hill; 2001:pp969-76.
- Gordon DS, Jones BM,
Browning SW, Spira TJ, Lawrence DN. Persistent polyclonal lymphocytosis
of B lymphocytes. N Engl J Med 1982;307:232-6. https://doi.org/10.1056/NEJM198207223070407 PMid:6979709

- Troussard
X, Valensi F, Debert C, Maynadie M, Schillinger F, Bonnet P, Macintyre
EA, Flandrin G. Persistent polyclonal lymphocytosis with binucleated B
lymphocytes: a genetic predisposition. Br J Haematol 1994;88:275-80. https://doi.org/10.1111/j.1365-2141.1994.tb05018.x PMid:7803270
- Mossafa
H, Malaure H, Maynadie M, Valensi F, Schillinger F, Garand R, Jung G,
Flandrin G, Troussard X. Persistent polyclonal B lymphocytosis with
binucleated lymphocytes: a study of 25 cases. Groupe Français
d'Hématologie Cellulaire. Br J Haematol 1999;104:486-93. https://doi.org/10.1046/j.1365-2141.1999.01200.x PMid:10086784
- Schmidt-Hieber
M, Burmeister T, Weimann A, Nagorsen D, Hofmann WK, Thiel E, Schwartz
S. Combined automated cell and flow cytometric analysis enables
recognition of persistent polyclonal B-cell lymphocytosis (PPBL), a
study of 25 patients. Ann Hematol 2008;87:829-36. https://doi.org/10.1007/s00277-008-0529-1 PMid:18587574
- Cornet
E, Lesesve JF, Mossafa H, Sébahoun G, Levy V, Davi F, Troussard X;
Groupe Français d'Hématologie Cellulaire (GFHC). Long-term follow-up of
111 patients with persistent polyclonal B-cell lymphocytosis with
binucleated lymphocytes. Leukemia 2009;23:419-22. https://doi.org/10.1038/leu.2008.208 PMid:18668130 PMCid:PMC2685812
- Matsue
K, Nishi H, Onozawa S, Itoh M, Tsukuda K, Yamaguchi M, Nakao S,
Kashimura M. Polyclonal B cell chronic lymphoproliferative disease with
hairy cell morphology: a case report and clonal studies. Am J Hematol
1996;51:141-6. https://doi.org/10.1002/(SICI)1096-8652(199602)51:2<141::AID-AJH8>3.0.CO;2-Y
- Machii
T, Yamaguchi M, Inoue R, Tokumine Y, Kuratsune H, Nagai H, Fukuda S,
Furuyama K, Yamada O, Yahata Y, Kitani T. Polyclonal B-cell
lymphocytosis with features resembling hairy cell leukemia-Japanese
variant. Blood 1997;89:2008-14. PMid:9058722
- Troussard
X, Mossafa H, Flandrin G. Identity between hairy B-cell
lymphoproliferative disorder and persistent polyclonal B lymphocytosis?
Blood 1997;90:2110-3. PMid:9292552
- Kanbayashi
H, Nagata K, Tanaka T, Matsuda S, Sakuma H, Maruyama Y, Machii T.
Polyclonal B-cell lymphocytosis with clinical and hematological
features resembling hairy cell leukemia. Jpn J Clin Hematol
1998;39:493-8 in Japanese.
- Yagi
Y, Sakabe H, Kaninoki R, Yoshikawa K, Inoue T, Fujiyama Y, Machii T. A
hairy B-cell lymphoproliferative disorder resembling hairy cell
leukemia. Jpn J Clin Hematol 2004;45:312-5 in Japanese.
- Nakahashi
H, Hashimoto Y, Yamane A, Irisawa H, Yokohama A, Saitoh T, Hanada H,
Matsushima T, Tsukamoto N, Karasawa M, Murakamai H, Nojima Y.
Polyclonal B-cell lymphocytosis with hairy cell appearance: hairy
B-cell lymphoproliferative disorder. Jpn J Clin Hematol 2007;48:647-51
in Japanese.
- Takeshita
T, Owatari S, Otsuka M, Hanada S. Remarkable response to cladribine in
the treatment for hairy B cell lymphoproliferative disorder. J Jpn Soc
Int Med 2007;96:1712-14 in Japanese.
- Okamoto
A, Inaba T, Fujita N. The role of interleukin-6 in a patient with
polyclonal hairy B-cell lymphoproliferative disorder: a case report.
Laboratory Hematology 2007;13:124-7. https://doi.org/10.1532/LH96.07015 PMid:18192143
- van
Dongen JJ, Langerak AW, Brüggemann M, Evans PA, Hummel M, Lavender FL,
Delabesse E, Davi F, Schuuring E, García-Sanz R, van Krieken JH, Droese
J, González D, Bastard C, White HE, Spaargaren M, González M, Parreira
A, Smith JL, Morgan GJ, Kneba M, Macintyre EA. Design and
standardization of PCR primers and protocols for detection of clonal
immunoglobulin and T-cell receptor gene recombinations in suspect
lymphoproliferations: report of the BIOMED-2 Concerted Action
BMH4-CT98-3936. Leukemia 2003;17:2257-317. https://doi.org/10.1038/sj.leu.2403202 PMid:14671650
- Tiacci
E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini
A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci
R, Elliott O, Liso A, Ambrosetti A, Pulsoni A, Forconi F, Trentin
L, Semenzato G, Inghirami G, Capponi M, Di Raimondo F, Patti C,
Arcaini L, Musto P, Pileri S, Haferlach C, Schnittger S, Pizzolo G, Foà
R, Farinelli L, Haferlach T, Pasqualucci L, Rabadan R, Falini B. BRAF
mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305-15. https://doi.org/10.1056/NEJMoa1014209 PMid:21663470 PMCid:PMC3689585
- Katayama
I, Mochino T, Honma T, Fukuda M. Hairy cell leukemia: a comparative
study of Japanese and non-Japanese patients. Semin Oncol 1984;11(4
Suppl. 2) 486-92. PMid:6505710
- Machii
T, Tokumine Y, Inoue R, Kitani T, Predominance of a distinct subtype of
hairy cell leukemia in Japan. Leukemia 1993;7:181-6.
PMid:8426471
- Delage
R, Roy J, Jacques L, Bernier V, Delâge JM, Darveau A. Multiple bcl-2/Ig
gene rearrangements in persistent polyclonal B-cell lymphocytosis. Br J
Haematol 1997;97:589-95. https://doi.org/10.1046/j.1365-2141.1997.852725.x PMid:9207405
- Chan
MA, Benedict SH, Carstairs KC, Francombe WH, Gelfand EW. Expansion of B
lymphocytes with an unusual immunoglobulin rearrangement associated
with atypical lymphocytosis and cigarette smoking. Am J Respair Cell
Mol Biol 1990;2:549-52. https://doi.org/10.1165/ajrcmb/2.6.549 PMid:2346660
[TOP]