Monia Ouederni1,2, Monia Ben Khaled1,2, Samia Rekaya1,2, Ilhem Ben Fraj1,2, Fethi Mellouli1,2 and Mohamed Bejaoui1,2
1 Pediatric Immuno-hematology unit, bone marrow transplantation center Tunis
2 Faculty of Medicine, University of Tunis El Manar, Tunisia
Corresponding
author: Monia Ouederni, Centre National de Greffe de Moelle Osseuse de
Tunis, Bab Saadoun, 2 Rue Jbel lakhdar, 1006 Tunis. Tel: 00 216 22 16
16 89, Fax: 00 216 71 56 53 68. E-mail:
moniahasan@yahoo.fr
Published: October 16, 2017
Received: June 19, 2017
Accepted: October 4, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017057 DOI
10.4084/MJHID.2017.057
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Hemophagocytic
lymphohistiocytosis (HLH) is a life-threatening
hyperinflammation caused by uncontrolled proliferation of
activated lymphocytes and histiocytes. Often, HLH is an acquired
syndrome. We report a case of a nine month-old-boy presented with
hepatosplenomegaly, severe anemia, thrombocytopenia,
hypertriglyceridemia and high hyperferritinemia. These clinical
features of HLH prompted a wide range of infectious and auto-immune
tests to be performed. After an extensive diagnostic workup, he
was referred to the immune-hematologic unit for HLH suspicion with an
unknown cause. Primary HLH due to familial lymphohistiocytosis
(FLH) was first evoked in front of consanguinity, probable HLH in the
family, early onset, and in the absence of a causative pathology like
infection or cancer. However, functional tests were normal. Atypical
features like the: absence of fever, hypotonia, recurrent diarrhea
since diversification, hematuria, and proteinuria suggested an inborn
metabolism error with gastrointestinal involvement. Specific tests were
performed to reach a final diagnosis.
|
Introduction
Hemophagocytic
lymphohistiocytosis (HLH) is a life-threatening hyperinflammation
caused by uncontrolled proliferation of activated lymphocytes and
histiocytes.[1] The diagnosis of HLH is challenging in patients with
prolonged fever, unresponsive to antibiotics. In 1994 the Histiocyte
Society defined a set of diagnostic criteria; they were
subsequently revised in 2004. The diagnosis of HLH can be established
either by molecular diagnosis consistent with HLH and/or in presence of
5/8 clinical and laboratory criteria for HLH: fever, splenomegaly,
cytopenia (affecting ≥2 of 3 lineages in peripheral blood),
hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis in
bone marrow or spleen or lymph nodes, low or absent NK cell activity,
ferritin ≥500µg/l, soluble CD25 (soluble IL-2 receptor) ≥2,400 U/ml.
Other supportive evidence includes cerebral symptoms with
moderate pleocytosis and/or elevated protein, elevated
transaminases and bilirubin, LDH. All features of HLH can be explained
by high concentrations of inflammatory cytokines and organ infiltration
by activated lymphocytes and histiocytes.[2,3]
HLH
can be primitive in children, underlying inherited immune deficiencies.
Primary HLH is an autosomal recessive or X-linked primary immune
deficiency including familial HLH (FLH) in which the clinical syndrome
of HLH is the only manifestation. Four subtypes of FLH are defined by
mutations in the following genes: PRF1 in FHL2, UNC13D in FHL3, STX11
in FHL4, and STXBP2 in FHL5. The Chediak-Higashi syndrome (CHS 1),
Griscelli syndrome (GS 2), Hermansky–Pudlak syndrome (HPS) and X linked
proliferative syndrome (XLP) are primary immune deficiencies having
distinctive clinical features besides the recurrent primary HLH.[2-4]
However,
HLH is, often, an acquired or secondary syndrome which can occur in all
age groups. Infection-associated HLH could be triggered by various
agents such as viruses of the herpes group, especially Epstein-Barr
virus (EBV) and cytomegalovirus (CMV) or by no viral agents such as
Leishmania. Acquired HLH could also be associated with malignant
diseases, especially lymphomas and to autoimmune diseases.[2-5]
It has rarely been described HLH secondary to inborn errors of
metabolism such as Lysinuric protein intolerance (LPI), a rare
metabolic disorder resulting from recessive-inherited mutations in the
SLC7A7 gene encoding the cationic amino-acids transporter subunit
y+LAT1. This pathology is characterized by protein-rich food
intolerance with secondary urea cycle disorder. It is a multiorgan
disease, that could lead to infiltrative lung disease, kidney failure
or auto-immune complications. The phenotypic heterogeneity of LPI has
resulted in various misdiagnoses.[6-11]
We report, herein, the
case of 9-month-old boy investigated for a persistent HLH with very
high hyperferritinemia. Throughout this case, we describe the
atypical presentation and outcome of HLH and we insist on differential
diagnosis of chronic HLH that must be kept in mind of specialists.
Report of the Case
Case presentation and clinical history.
M.K is a 9-month-old boy, born from a consanguineous marriage. He had
been breastfed for seven months with normal growth. Since food
diversification, he exhibited poor weight gain, developed recurrent
diarrhea, hepatomegaly, and splenomegaly with pancytopenia,
increased serum ferritin and lactate dehydrogenase (LDH) level. Hence,
he was referred to the immune-hematologic unit, for
hemophagocytic lymphohistiocytosis (HLH) suspicion.
Initial workup.
Physical examination showed pallor, hypotonia, failure to thrive, liver
and spleen enlargement. No fever was noted. The physical examination
did not reveal other abnormalities. Urine bandlets showed
proteinuria and microscopic hematuria. Laboratory findings (Table 1)
showed pancytopenia with normochromic normocytic non- regenerative
anemia; neutropenia, lymphopenia, and thrombocytopenia),
hyperferritinemia (8000 ng/ml), elevated
triglycerides (14 mmol/l), high cholesterol (8 mmol/l) (Figure 1),
elevated very low-density lipoproteins (VLDL) and low high-density
lipoproteins (HDL). He had low fibrinogen (0.74 g/l), without other
signs of disseminated intravascular coagulation, increased LDH (3200
UI/l), low urea (1,29 mmol/l) and normal creatinine. Other routine
biological tests were normal. Blood and bone marrow smears showed no
hemophagocytosis. Cerebrospinal fluid (CSF) exam showed no activated
cells. Cerebral MRI was normal.
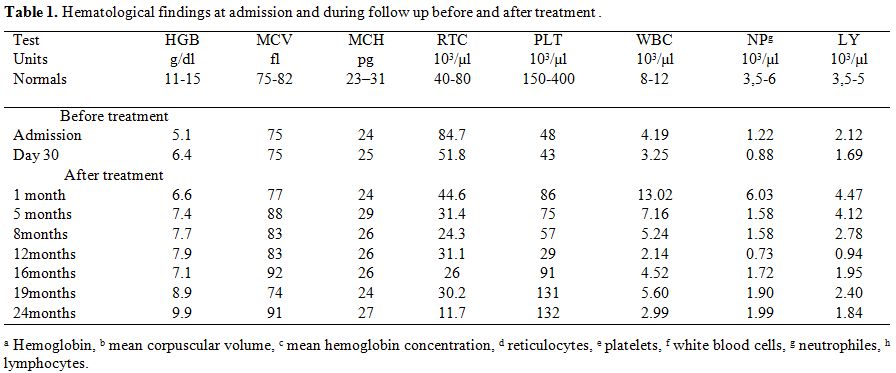 |
Table 1.
Hematological findings at admission and during follow up before and after treatment. |
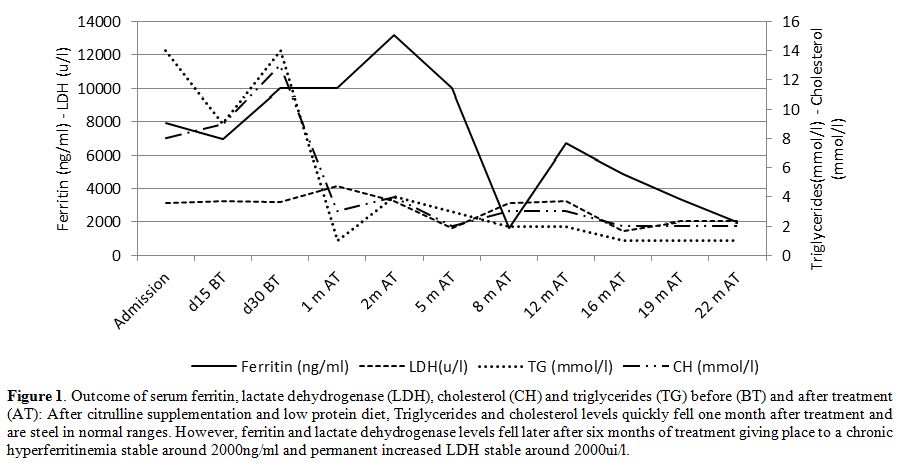 |
Figure 1. Outcome of serum ferritin,
lactate dehydrogenase (LDH), cholesterol (CH) and triglycerides (TG)
before (BT) and after treatment (AT): After citrulline supplementation
and low protein diet, Triglycerides and cholesterol levels quickly fell
one month after treatment and are steel in normal ranges. However,
ferritin and lactate dehydrogenase levels fell later after six months
of treatment giving place to a chronic hyperferritinemia stable around
2000ng/ml and permanent increased LDH stable around 2000ui/l. |
Differential diagnosis and further investigations.
These clinical features of Hemophagocytic lymphohistiocytosis (HLH)
prompted performing other investigations looking for an acquired HLH.
So a complete workup was made to rule out an infection,
an autoimmune disease or malignancy: microbiological and
autoimmunity tests were negative, and blood and bone marrow smears
showed no blasts.
Differential diagnosis and further investigations.
A primary HLH was suspected. The patient had no albinism suggesting a
GS 2 or a CHS 1. He had no EBV infection suggesting an XLP. FLH was
thought to be the primary cause of HLH due to parental consanguinity,
history of a cousin with the same signs died at the age of 4 months, in
the absence of other evident causes. Nevertheless, perforin expression
and degranulation test were regular. Other underlying primary immune
deficiency was searched: Immunologic tests showed average
immunoglobulin G, A and M levels, moderate global lymphopenia CD4, CD8,
B and NK and no increased activated lymphocytes (Table 2).
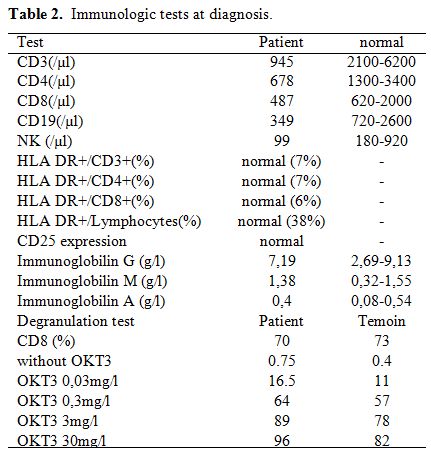 |
Table 2. Immunologic tests at diagnosis. |
Final diagnosis.
Moreover, several atypical elements were noted in this HLH: absence of
fever, hypotonia without neurological activation detected on the CSF
exam and cerebral MRI, recurrent diarrhea, hematuria and proteinuria,
very high cholesterol level, and not increased HLA-DR expression. All
these manifestations beginning since food diversification oriented to
lysinuric protein intolerance (LPI). In fact, other metabolic errors
mimicking HLH similarly like lysosomal acid lipase deficiency, or
galactosemia manifest in the first days of life or at lactation, and
Gaucher disease was excluded by absence of the typical cells in bone
marrow and by a regular glucocerebrosidase activity in cultured
fibroblasts.
Serum ammonium level was found increased at 112UI/l
(normal ≤70UI/l). Metabolic tests showed an increased urinary excretion
of orotic acid. The amino acid analyses from plasma and urine showed
low plasma levels of cationic amino acids (CAAs) and increased urinary
excretion of CAAs. Organic - acid analyses from urine at diagnosis
showed increased urinary intermediary organic acids of the Krebs cycle (Table 3).
This profile with low plasma levels of CAAs and increased urinary
excretion, orotic aciduria and hyperammonemia is compatible with a
defect in the y+LAT1 sub-unit of the cationic amino-acids transporter
encoded by the SLC7A7 gene.
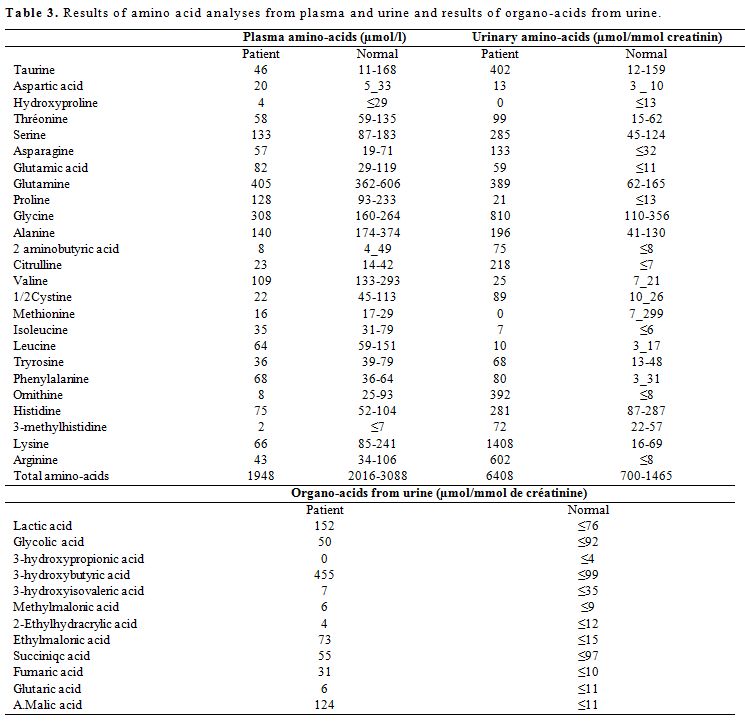 |
Table 3. Results of amino acid analyses from plasma and urine and results of organo-acids from urine. |
Treatment and outcome.
Citrulin supplementation to 100 mg/kg/day was prescribed with protein
intake limited to 0.8 g/kg/day. Liver and spleen enlargement decreased,
hypotonia disappeared. The patient has gained 12 kg in two years. He
needed repeated platelet and blood cell transfusions during the first
month. Hematological disorders have gradually improved with persistent
mild anemia not requiring further transfusions (Table 1).
Ammonium, triglycerides, and cholesterol quickly fell to normal levels
one month after treatment and are still in average ranges. Fibrinogen
value normalized within 5 months. Serum ferritin and LDH fell later
after six months giving place to a persistent hyperferritinemia around
2000 ng/ml and permanent increased LDH around 2000 UI/l (Figure 1). During follow up, 24 months of treatment, no further authentic HLH occurred.
Discussion
Hemophagocytic
lymphohistiocytosis (HLH) is a life-threatening hyperinflammation
caused by uncontrolled proliferation of activated lymphocytes and
histiocytes.[1] Often, HLH is an acquired syndrome.[5] However, HLH can
be primitive in children, underlying inherited immune deficiencies.[3,4]
An additional HLH cause, the hereditary metabolic diseases and
especially HLH related to lysinuric protein intolerance (LPI), is more
and more
described.[6-9] The phenotypic heterogeneity of LPI
has resulted in various misdiagnoses, of the most frequent is familial
lymphohistiocytosis (FLH) given clinical features of HLH which are also
often found in LPI.[10-12]
According to revised haemophagocytic
lymphohistiocytosis (HLH) 2004 diagnostic criteria our patient fulfills
only four of eight criteria. So, the diagnosis of leaky HLH was kept.
Other laboratory findings, which are considered to be of diagnostic
value in HLH, were also identified in our patient: hepatic enzyme
abnormalities, elevated LDH, elevated VLDL and low HDL. In fact, in
these revisited criteria haemophagocytosis in bone marrow aspirate is
not constant.[1,2,3] Moreover, some patients enrolled in the
International Registry of HLH do not fulfill all the diagnostic
criteria.[13] It is currently accepted that hyperferritinemia in LPI is
associated with LPI-related HLH even though the other HLH criteria are
not prominent.[13] Therefore, most of the non-metabolic biomarkers of
LPI are explained by an underlying chronic or quiescent HLH that can
progress to active HLH with fever.[14] The hepatosplenomegaly observed
in LPI reflect HLH rather than the nutritional depletion of CAAs.[15]
HLH related LPI described in our patient differs from HLH in FLH. In
LPI, HLH is chronic and intermittent. The hyperferritinemia and high
LDH are usually the only permanent findings.[13-15] Our patient has
never normalized his ferritin and LDH levels. However, in FLH all those
abnormalities are reversible and normalized when patients went into
remission.[1,2] Then, fever, the most constant feature and typically
prolonged in FLH related HLH,[1] was absent in our patient. It has been
described that fever is not a prominent finding in LPI related HLH.[7] We
suggest that patients with suspected HLH, based on the clinical
syndrome, should undergo the functional screening that is now a
standing point in the diagnosis of HLH and FLH. Based on those
findings, the lack of any functional defect could be one more strong
argument to evaluate LPI in a child with no fever, growth retardation,
and lack of the typical dysfunction of FLH. Distinguishing features
between FLH related HLH and LPI related HLH are summarized in Table 4.
The phenotypic heterogeneity of LPI has resulted in various other
misdiagnoses reported in the literature such as cases of LPI
misdiagnosed as food protein-induced enterocolitis syndrome.[16] The
diagnosis of LPI was also made in a 5-year-old male child followed for
3 years for multiple fractures, idiopathic osteoporosis, and short
stature in the absence of typical features of LPI.[17] These unusual
presentations are responsible for diagnosis delay of rare disorders for
which early intervention may modify the clinical course.
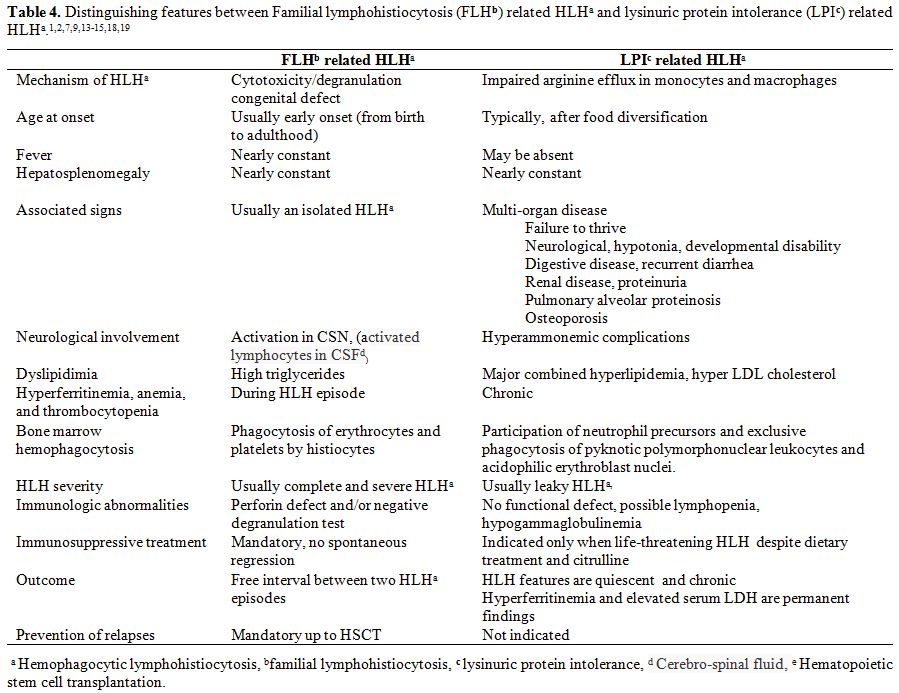 |
Table 4. Distinguishing features between
Familial lymphohistiocytosis (FLHb) related HLHa and lysinuric protein
intolerance (LPIc) related HLHa.[1-2,7,9,13-15,18-19] |
Our
patient, like most of LPI subjects, displayed, other hematological and
immunological abnormalities including chronic and intermittent anemia,
thrombocytopenia, neutropenia and moderate global lymphopenia. Signs of
T-cells dysfunction are usually present whenever investigated in
LPI.[7,14,15]
The most evocative element of LPI in our patient,
calling into question the FLH, was the association of leaky HLH to
other features such as failure to thrive, extreme hyperlipidemia,
neurological and kidney involvement and the onset of manifestations
since food diversification.[9,11,14,15] Proteinuria and microscopic
hematuria should be followed over time since it can develop Fanconi
syndrome or end-stage renal disease requiring dialysis.[8,11]
The
diagnosis of LPI is based on the presence of, at least, four of the
following findings:[6,11] (1) low plasma levels of CAAs; (2) increased
urinary excretion of CAAs; (3) orotic aciduria; (4) hyperammonemia
generally mild with usual protein intakes, prevented by oral
administration of citrulline; and (5) reduced intestinal absorption of
CAAs after an oral loading test. The first four criteria were present
in our patient. Nutritional imbalance of CAAs does not explain the
aberrant inflammatory and immune responses.[15] The mutation of SLC7A7
gene strongly impairs arginine efflux through system y+L in LPI
monocytes and macrophages. It has been suggested that this may have a
role in the crosstalk between T lymphocytes and macrophage leading to a
defect in lymphocyte cytotoxic activity that prevents the efficient
removal of antigens and results in abnormal immune activation of CTLs
and macrophages explaining HLH in LPI.[18,19]
There have
been rare case reports of HLH secondary to other inborn errors of
metabolism. HLH was described in Wolman disease, a severe systemic
disease manifesting in the first days of life with vomiting, diarrhea,
failure to thrive, hepatosplenomegaly, jaundice, anemia, and
thrombocytopenia. A neonatal onset with distinctive markers of the
disease such as subcapsular adrenal calcification and the presence of
cytoplasmic lipid-laden vacuoles on bone marrow smear indicate an
assessment of leukocytic cholesteryl esterase activity on blood
leukocytes.[20,21] Biotinidase deficiency should also be considered as
a differential diagnosis of patients fulfilling HLH criteria,
especially in the presence of ketolactic acidosis and organic
aciduria.[22]
Three cases with organic acidemia who developed HLH
during the course of metabolic disorder have been reported. All the
patients presented with metabolic acidosis and ketosis and increased
histiocytes, lipid-laden macrophages in bone marrow aspirate.[23] It
was reported a case of an infant with early-onset cobalamin C
deficiency who presented with HLH with symptoms of feeding difficulty,
hypotonia, lethargy, and seizures in the first month of life. Urine
organic acid analysis, acylcarnitine profile, and plasma homocysteine
could orient to the diagnosis which must be confirmed by specific
tests.[24] HLH has also been described in association with Gaucher
disease. Features of Gaucher disease, which are common to HLH, include
unexplained fevers and cytopenias, both of which are explainable by the
inflammation mediated by macrophages.[25] In our patient, the cultured
fibroblasts enzyme assay revealed normal glucocerebrosidase activity.
These cases suggest that a careful metabolic workup should be
performed, extending to more advanced tests than organic and amino-acid
analyzes when facing to a pediatric patient with HLH especially if
clinical features of the patient suggested a metabolic disorder
including hypotonia, irritability, or mild developmental delay.
The
persistent symptoms mimicking HLH in our patient must be carefully
monitored since it can progress to a life-threatening condition.
Immunosuppressive drugs should be considered in LPI only when there is
a clear threat to life.[9,14] It was reported that combined
hyperlipidemia frequently seen in LPI requires a specific treatment
with HMG-CoA reductase inhibitors.[26] However, hyperlipidemia
disappeared quickly in our patient. Citrulline treatment does not
improve all features in our patient. Large amounts of citrulline
increase the intracellular synthesis of arginine and may further
stimulate the immune cascade in reticular endothelial cells.[15] It has
been suggested that lysine supplementation could be able to ameliorate
the clinical symptoms of LPI that are not corrected by citrulline.[27]
In
a child presenting HLH, a wide range of exams should be performed to
rule out an infection, an autoimmune disease or malignancy, since most
of these causes are treatable. If primary HLH is suspected, an
underlying immune deficiency like FLH, GS, CHS, XLP, should be
screened. However, metabolic diseases such as LPI must be kept in mind
of specialists as a differential diagnosis of HLH, and a careful
metabolic workup should be performed when facing to a pediatric patient
with HLH especially if clinical features suggested a metabolic
disorder. The lysinuric protein intolerance should be considered in the
differential diagnosis of familial lymphohistiocytosis, especially in
the absence of fever and the association of atypical clinical and
biological features to HLH including hypotonia, irritability, food
intolerance, and renal involvement. We suggest that patients with
suspected HLH, based on the clinical syndrome, should undergo the
functional screening. The lack of any functional defect could be one
more strong argument to evaluate LPI in a child with no fever, growth
retardation, and lack of the typical dysfunction of FLH. An early
diagnosis of LPI can prevent unnecessary intensive immunosuppressive
therapy and bone marrow transplantation. LPI related HLH is often
chronic and quiescent with permanent hyperferritinemia. However, it
must be carefully monitored since it can progress to life-threatening
HLH.
References
- Gholam C, Grigoriadou S, Gilmour K.C, Gaspar H.B.
Familial haemophagocytic lymphohistiocytosis: advances in the genetic
basis, diagnosis and management. Clinical and Experimental Immunology
2011; 163: 271-83. https://doi.org/10.1111/j.1365-2249.2010.04302.x
PMid:21303357 PMCid:PMC3048610

- Ramachandran
S, Zaidi F, Aggarwal A, Gera R. Recent advances in diagnostic and
therapeutic guidelines for primary and secondary hemophagocytic
lymphohistiocytosis. Blood Cells Mol Dis 2017; 64: 53–57.
https://doi.org/10.1016/j.bcmd.2016.10.023 PMid:28433836
- Janka
G.E. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J
Pediatr 2007; 166 (2):95–109 https://doi.org/10.1007/s00431-006-0258-1
PMid:17151879
- Imashuku
S, Ueda I, Teramura T, Mori K, Morimoto A, Sako M, Ishii E. Occurrence
of haemophagocytic lymphohistiocytosis at less than 1 year of age:
analysis of 96 patients. Eur J Pediatr 2005; 164 (5): 315–19.
https://doi.org/10.1007/s00431-005-1636-9 PMid:15731905
- Mehta
RS, Smith RE. Hemophagocytic lymphohistiocytosis (HLH): a review of
literature. Med Oncol 2013; 30(4):740.
https://doi.org/10.1007/s12032-013-0740-3 PMid:24105023
- Parenti
G, Sebastio G, Strisciuglio P, Incerti B, Pecoraro C, Terracciano L,
Andria G. Lysinuric protein intolerance characterized by bone marrow
abnormalities and severe clinical course. J. Pediatr 1995; 126
(2):246–51. https://doi.org/10.1016/S0022-3476(95)70552-X
- Duval
M, Fenneteau O, Doireau V, Faye A, Emilie D, Yotnda P, Drapier JC,
Schlegel N, Sterkers G, de Baulny HO, Vilmer E. Intermittent
hemophagocytic lymphohistiocytosis is a regular feature of lysinuric
protein intolerance. J Pediatr 1999; 134 (2):236-9.
https://doi.org/10.1016/S0022-3476(99)70423-3
- Tanner
L.M, Nanto-Salonen K, Niinikoski H, Jahnukainen T, Keskinen P, Saha H,
Kananen K, Helanterä A, Metso M, Linnanvuo M, Huoponen K, Simell O.
Nephropathy advancing to end-stage renal disease: a novel complication
of lysinuric protein intolerance. J. Pediatr 2007; 150 (6):631–34.
https://doi.org/10.1016/j.jpeds.2007.01.043 PMid:17517249
- Güzel-Ozantürk
A, Ozgül RK, Unal O, Hişmi B, Aydın Hİ, Sivri S, Tokatlı A, Coşkun T,
Aksöz E, Dursun A. Molecular and clinical evaluation of Turkish
patients with lysinuric protein intolerance. Gene 2013;521 (2): 293–5.
https://doi.org/10.1016/j.gene.2013.03.033 PMid:23542076
- Gupta
S, Weitzman S. Primary and secondary hemophagocytic
lymphohistiocytosis: clinical features, pathogenesis and therapy.
Expert Rev Clin Immunol 2010; 6 (1): 137–54.
https://doi.org/10.1586/eci.09.58 PMid:20383897
- Mauhin
W, Habarou F, Gobin S, Servais A, Brassier A, Grisel C, Roda C, Pinto
G, Moshous D, Ghalim F, Krug P, Deltour N, Pontoizeau C, Dubois S,
Assoun M, Galmiche L, Bonnefont JP, Ottolenghi C, de Blic J, Arnoux JB,
de Lonlay P. Update on Lysinuric Protein Intolerance, a Multi-faceted
Disease Retrospective cohort analysis from birth to adulthood. Orphanet
J Rare Dis 2017 ; 12(1):3. https://doi.org/10.1186/s13023-016-0550-8
PMid:28057010 PMCid:PMC5217205
- Shinsaku
I. Hyperferritinemia in Hemophagocytic Lymphohistiocytosis and Related
Diseases. Pediatr Blood Cancer 2008; 51 (3):442–6
https://doi.org/10.1002/pbc.21623 PMid:18506755
- Aricò
M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M,
Rusca MP. Hemophagocytic lymphohistiocytosis. Report of 122 children
from the International Registry. FHL Study Group of the Histiocyte
Society. Leukemia 1996; 10(2):197-203. PMid:8637226
- Ogier
de Baulny H, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance
(LPI): A multi-organ disease by far more complex than a classic urea
cycle disorder. Molecular Genetics and Metabolism 2012; 106 (1):12–7
https://doi.org/10.1016/j.ymgme.2012.02.010 PMid:22402328
- Sebastio
G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing
concepts on a multisystem disease. Am J Med Genet C Semin Med Genet
2011; 157C(1):54-62 https://doi.org/10.1002/ajmg.c.30287 PMid:21308987
- Maines
E, Comberiati P, Piacentini GL, Boner AL, Peroni DG. Lysinuric protein
intolerance can be misdiagnosed as food protein-induced enterocolitis
syndrome. Pediatric Allergy and Immunology 2013; 24: 509–510
https://doi.org/10.1111/pai.12096 PMid:23772603
- Posey
JE, Burrage LC, Miller MJ, Liu P, Hardison MT, Elsea SH, Sun Q, Yang Y,
Willis AS, Schlesinger AE, Bacino CA, Lee BH. Lysinuric Protein
Intolerance Presenting with Multiple Fractures. Mol Genet Metab Rep
2014; 1: 176–183 https://doi.org/10.1016/j.ymgmr.2014.03.004
PMid:25419514 PMCid:PMC4235665
- Barilli
A, Rotoli BM, Visigalli R, Bussolati O, Gazzola GC,Gatti R,
Dionisi-Vici C, Martinelli D, Goffredo BM, Font-Llitjós M, Mariani F,
Luisetti M, Dall'Asta V. Impaired phagocytosis in macrophages from
patients affected by lysinuric protein intolerance. Mol Genet Metab
2012; 105(4):585-9 https://doi.org/10.1016/j.ymgme.2012.01.008
PMid:22325938
- Barilli
A, Rotoli B.M, Visigalli R, Bussolati O, Gazzola G.C, Kadija Z, Rodi G,
Mariani F, Ruzza ML, Luisetti M, Dall'Asta V. In lysinuric protein
intolerance system y+L activity is defective in monocytes and in
GM-CSF-differentiated macrophages. Orphanet J Rare Dis 2010; 5:32. https://doi.org/10.1186/1750-1172-5-32 PMid:21110863 PMCid:PMC2999609
- Rabah
F, Al-Hashmi N, Beshlawi I. Wolman's disease with secondary
hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol 2014;
31(6):576-8. https://doi.org/10.3109/08880018.2014.920942 PMid:24933302
- Taurisano
R, Maiorana A, De Benedetti F, Dionisi-Vici C, Boldrini R, Deodato F.
Wolman disease associated with hemophagocytic lymphohistiocytosis:
attempts for an explanation. Eur J Pediatr 2014;173(10):1391-4. https://doi.org/10.1007/s00431-014-2338-y PMid:24844354
- Kardas
F, Patiroglu T, Unal E, Chiang SC, Bryceson YT, Kendirci M.
Hemophagocytic syndrome in a 4-month-old infant with biotinidase
deficiency. Pediatr Blood Cancer 2012; 59(1):191-3. https://doi.org/10.1002/pbc.23247 PMid:22605457
- Gokce
M, Unal O, Hismi B, Gumruk F, Coskun T, Balta G, Unal S, Cetin M,
Kalkanoglu-Sivri HS, Dursun A, Tokatlı A. Secondary hemophagocytosis in
3 patients with organic acidemia involving propionate metabolism.
Pediatr Hematol Oncol 2012; 29(1) :92-8. https://doi.org/10.3109/08880018.2011.601402 PMid:21970506
- Susan
Wu, Ignacio Gonzalez-Gomez, Thomas Coates, Shoji Yano. Cobalamin C
disease presenting with hemophagocytic lymphohistiocytosis. Pediatr
Hematol Oncol 2005; 22(8):717-21. https://doi.org/10.1080/08880010500278871 PMid:16251179
- Sharpe
LR, Ancliff P, Amrolia P, Gilmour KC, Vellodi A. Type II Gaucher
disease manifesting as haemophagocytic lymphohistiocytosis. J Inherit
Metab Dis. 2009; 32 (1): S107-10. https://doi.org/10.1007/s10545-009-1091-2 PMid:19267217
- Tanner
LM, Niinikoski H, Näntö-Salonen K, Simell O. Combined hyperlipidemia in
patients with lysinuric protein intolerance. J Inherit Metab Dis 2010;
33 Suppl 3:S145-50. https://doi.org/10.1007/s10545-010-9050-5 PMid:20177788
- Lukkarinen
M, Nanto-Salonen K, Pulkki K, Aalto M, Simel O. Oral Supplementation
Corrects Plasma Lysine Concentrations in Lysinuric Protein Intolerance.
Metabolism 2003; 52(7): 935-8. https://doi.org/10.1016/S0026-0495(03)00089-1
P]