Oluwagbemiga O. Adeodu1, Morenike A. Akinlosotu2, Samuel A. Adegoke1 and Saheed B.A. Oseni1
1 Department of Paediatrics and Child Health, Obafemi Awolowo University, Ile Ife, Osun State, Nigeria.
2 Department of Paediatrics, Obafemi Awolowo University Teaching Hospitals Complex, Ile Ife, Osun State, Nigeria.
Corresponding
author: Samuel A. Adegoke, Department of Paediatrics and Child Health,
Obafemi Awolowo University, Ile-Ife, Nigeria, P.M.B. 013, Ile-Ife,
Nigeria. E-mail:
adegoke2samade@yahoo.com
Published: November 1, 2017
Received: July 17, 2017
Accepted: September 19, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017063 DOI
10.4084/MJHID.2017.063
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background: Foetal
haemoglobin (HbF) is a major modifying factor influencing sickle cell
disease (SCD) severity. Despite this, HbF estimation is not routinely
done in Nigeria. The relationship between HbF and SCD severity among
affected children is also poorly studied.
Methods:
In this descriptive cross-sectional study, we determined the
relationship between steady state HbF levels and disease severity of
Nigerian children aged 1 – 15 years with homozygous SCD. For each
child, the socio-demographic characteristics and SCD clinical severity
were determined. The latter was assessed based on the frequency of
significant painful episodes, blood transfusion, and hospitalisation in
the preceding 12 months; lifetime cumulative incidence of SCD-related
complications; the degree of splenic and hepatic enlargement; current
haematocrit and leucocyte count. Foetal haemoglobin levels were
quantified with high-performance liquid chromatography.
Results:
The mean HbF level of the 105 children with SCA was 9.9 ± 6.0%. Male
had significantly lower mean HbF levels than females, 8.0 ± 5.6% vs.
12.2 ± 5.8% (p < 0.001). None of the children had severe disease.
However, the 32 children with moderate disease had significantly lower
mean foetal haemoglobin levels than the 73 with mild disease (7.7 ±
5.6% vs 10.8 ± 6.0% respectively). The mean HbF level was also
significantly lower in children who had a history of acute chest
syndrome and stroke compared to those without these complications, p =
0.002 and 0.010 respectively.
Conclusion: Children
with SCA who had a moderate disease and those with a history of
life-threatening complications such as stroke and acute chest syndrome
had significantly low HbF levels. Therefore, it is recommended that
facilities for early quantification of foetal haemoglobin and HbF
inducement were made available to reduce the morbidity and mortality
among these children.
|
Introduction
Globally, sickle cell anaemia (SCA) is the most common inherited haematological disorder.[1]
It is found more frequently in sub-Saharan Africa, where it
significantly contributes to the morbidity and mortality among
children. It accounts for 5-16% of under-five mortality in the West
African sub-region.[2] Nigeria has the largest burden of sickle cell anaemia worldwide with about 150,000 affected babies being born annually.[2]
Affected children suffer varying morbidities contingent on the
availability of appropriate care, necessary tools and drugs needed for
management of the disease.
The presence of high foetal
haemoglobin had been documented to ameliorate the disease severity in
the western world where children with SCD are routinely given
hydroxyurea in order to induce the production of foetal haemoglobin and
reduce frequent crises.[3] However, children in
Nigeria and most other African countries still suffer devastating
complications such as stroke, priapism, acute chest syndrome and deep
sited infections like meningitis and cerebral abscess.[4]
Foetal haemoglobin level had been reported to be higher among Jamaican
and Asian children with the consequent milder course of the disease.[5]
In Nigeria, there is a paucity of data on the influence of foetal
haemoglobin levels on disease severity among children with SCA. Hence,
this study aimed at determining the relationship between foetal
haemoglobin level and disease severity among children with SCA in
steady state.
Methods
A
descriptive cross-sectional study was carried out in the Paediatric
sickle cell disease clinic of the Wesley Guild Hospital unit, Obafemi
Awolowo University Teaching Hospitals Complex, Ile Ife. A total of 105
children with SCA between age one and 15 years who were in steady state
(no crisis, infection or fever for at least four weeks and no blood
transfusion in the preceding three months) were consecutively enrolled.
Children with other haematological disorders such as
Glucose-6-Phosphate Dehydrogenase deficiency; and those with chronic
liver, kidney and heart diseases were excluded. Children on
hydroxyurea, those who did not give assent and those whose parent
refused consent were not included. The study was approved by the
Hospital Ethics/Research Committee (ERC/2013/11/12) and consent
obtained from each parent/caregiver and assent from the children as
appropriate.
A data proforma was used to obtain the
socio-demographic characteristics such as age, sex and socio-economic
class of participants as described by Oyedeji based on rank assessment
of parental occupation and the level of education.[6] The clinical severity of SCD was determined based on the number of admissions, blood transfusions and
significant painful crises (pain episode that requires a hospital visit and the use of analgesic)[7] in the preceding 12 months and other complications present as described by Adegoke et al.[8]
Patients with a score of less than 8 were classified as having mild
disease, 8 to 17 as moderate disease and greater than 17 as a severe
disease from a total obtainable score of 34.
Venous blood sample
was obtained and analysed for the complete blood count using ABX Micros
ES 60® automated haemoanalyser and foetal haemoglobin levels using an
automated BIO-RAD® D10 high-performance liquid chromatography (HPLC)
machine at the Haematology Laboratory of the National Sickle Cell
Foundation Lagos, Nigeria. Using the cut-off values of 10%, patients
with HbF levels <10% were categorised as having low HbF while those
with values ≥10% were categorised as having high HbF levels.[9]
Statistical analysis. Data were analysed using the statistical package for the social sciences (SPSS) software for windows version 17.0.[10]
Means (± standard deviation, SD), median, proportions and percentages
were determined as applicable. The means and standard deviations (±SD)
were calculated for continuous variables while proportions and
percentages were calculated for categorical variables. Categorical
variables were compared with chi-squared or Fisher’s exact tests while
continuous data were compared with independent sample t-test,
Mann-Whitney U test or Analysis of variance (ANOVA) as indicated. The
degree of correlation of continuous data was determined by Pearson’s
correlation analysis. Logistic regression analysis was done to examine
the independent effect of foetal haemoglobin on SCD severity.
Statistical significance was established when the p-value was less than
0.05.
Results
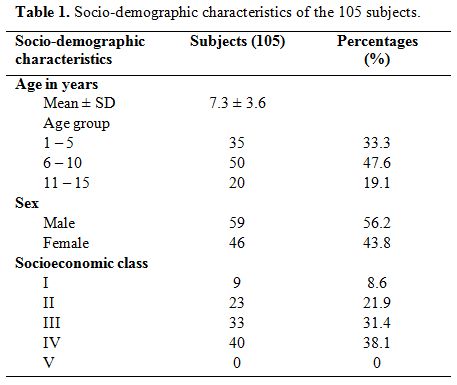
Of
the total 105 children with SCA studied, 59 (56.2%) were males with a
M: F of 1.3:1. Their ages ranged from one to 15 years with a mean of
7.3 ± 3.6 years. Thirty-five (33.3%) were preschool (1–5 years), 50
(47.6%) were children aged 6–10 years and 20 (19.1%) were adolescents
(>10 years). Forty (38.1%) of the population were from the lower
social class (classes 4 and 5) while the middle and upper classes
constituted 31.4% and 30.5% respectively. Table 1 shows the age, sex and socioeconomic class distribution of the subjects studied.
 |
Table
1. Socio-demographic characteristics of the 105 subjects. |
Clinical burden of the disease. Table 2
shows that 89 (84.8%) experienced at least one episode of pain and 33
(31.4%) had more than three significant painful episodes requiring
hospital visit and the use of analgesia in the 12 months preceding the
study. Sixteen (15.2%) did not experience any significant painful
episode in the year preceding the study. Fifty-one (48.6%) children
required hospitalisation including two (1.9%) that required more than
three admissions in the 12 months preceding the study. Twenty-one (20%)
children were transfused at least once in the year prior to
recruitment. Also, 45 (42.9%) subjects had a history of dactylitis. The
mean age at which they had the first dactylitis was 1.2±1.0 years,
ranging from three months to four years.
 |
Table 2. Frequency of pain episodes,
SCD-related hospitalisation and transfusion among the subjects in the
12 months preceding the study. |
Sickle cell disease severity.
The sickle cell disease severity score ranged from 0-14 with a mean
score of 6.0±3.2. Using a score of 0-7 for mild disease, 8-17 for
moderate and >17 for severe disease, 73 (69.5%) had mild disease, 32
(30.5%) had moderate disease while none had severe disease.
The
most common complication of SCA among the subjects was acute chest
syndrome, seen in 19 (18.1%) of the children followed by osteomyelitis
in 14 (13.3%). Other complications included stroke in 3 (2.9%),
avascular necrosis of head of femur, chronic leg ulcers and priapism, 2
(1.9%) each.
Foetal haemoglobin levels in the subjects.
The mean HbF level was 9.9 ± 6.0% with a range of 0.8 - 27.6%.
Sixty-six (62.9%) had low HbF levels of less than 10% while 39 (37.1%)
had high HbF value ≥10%. None had a 0% value.
Relationship between socio-demographic characteristics and foetal haemoglobin. Table 3
shows that male children had significantly lower mean haemoglobin F
levels than females, 8.0±5.6% vs 12.2±5.8% (p<0.001). Also,
significantly higher proportion of males (78.0% vs. 22.0% of females)
had HbF levels <10%; χ2=13.168;
p<0.001. Additionally, the mean HbF across the age groups decreased
with age and this was statistically significant (χ2=3.851,
p=0.024). The 39 subjects who had high HbF were significantly younger
than those with low HbF (6.3±4.1 years vs. 7.8±3.3 years, t=2.150,
p=0.034). Also, 19 (54.3%) of the 35 children who were 1-5 years
compared with 20 (28.6%) of the 70 who were >5 years (i.e. age
groups 6-10 years and 11-15 years) had high HbF levels, χ2=6.608, p=0.010. There was a significant inverse correlation between HbF levels and age for both sexes, as shown in figure 1, r=- 0.3, p=0.004 for females, and r=- 0.3 , p=0.006 for males.
On
the other hand, the mean HbF levels were not statistically different in
the socioeconomic classes (p=0.877) and were not influenced by the
frequency of pain crisis, blood transfusion and hospital admissions,
p=0.147; 0.808; and 0.347 respectively.
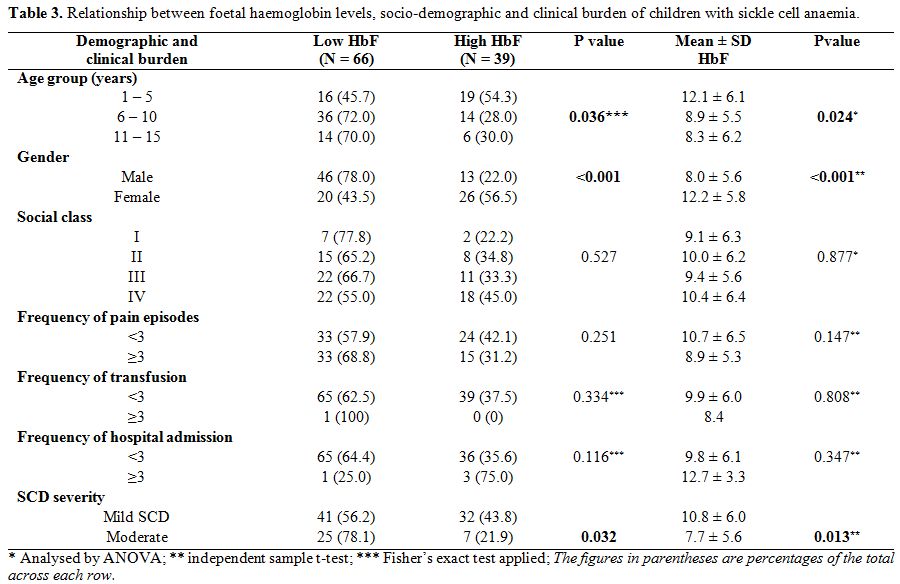 |
Table 3.
Relationship between foetal haemoglobin levels, socio-demographic
and clinical burden of children with sickle cell anaemia. |
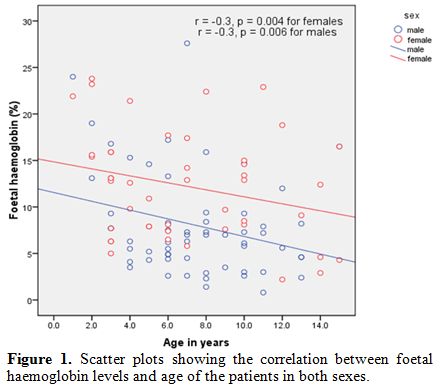 |
Figure 1. Scatter plots showing the correlation between foetal haemoglobin levels and age of the patients in both sexes. |
Relationship between socio-demographic characteristics and SCD severity.
Disease severity worsens with age. Higher proportion of those >5
years, i.e. 27 (38.6%) of the 70 as against 5 (14.3%) of the 35 aged
1-5 years had moderate SCD severity. Those older than 5 years were 3.8
times more likely to have moderate disease severity than those aged 1-5
years, odd ratio =3.8, 95% confidence interval =1.3-10.9, p=0.011.
Also, more males, 39.0% (23/59) compared to 19.6% females (9/46) had
moderate disease, OR=0.4, 95% CI=0.2-0.9, p=0.032. Socioeconomic class
did not significantly influence SCD severity. The proportions of
children with mild disease who came from high social class (66.6%) and
middle/low social classes (69.8%) were similar, OR=0.9, 95% CI=0.2-3.7,
p=1.000.
Relationship between foetal haemoglobin and disease severity.
Those with moderate disease had significantly lower mean foetal
haemoglobin levels than those with mild disease (7.7±5.6% vs 10.8±6.0%
respectively; p=0.013). Also, significantly higher proportion of
subjects with moderate disease (78.1%) as against those with mild
disease (56.2%) had HbF levels <10% (χ2=4.596,
p=0.032). On the other hand, more of the children with mild SCD
severity (43.8%) than those with moderate disease (21.9%) had high
levels of HbF (HbF ≥10%).Sickle cell disease severity score had
significant inverse correlation with HbF levels (r=- 0.3, p=0.002).
Relationship between foetal haemoglobin and clinical characteristics. Table 4 shows
that the mean foetal haemoglobin level was significantly lower in
children who had a history of acute chest syndrome than those without
this complication, (t=-3.243, p=0.002). Similarly, children with a
history of stroke had significantly lower HbF than those without the
history of stroke (t=- 4.296, p=0.010). There was, however, no
difference in the mean foetal haemoglobin levels of those with a
history of osteomyelitis, septic arthritis, leg ulcer, priapism,
avascular necrosis and dactylitis when compared to those without these
complications. From table 5, higher proportion of children with low HbF had previous ACS (χ2=5.033, p=0.025).
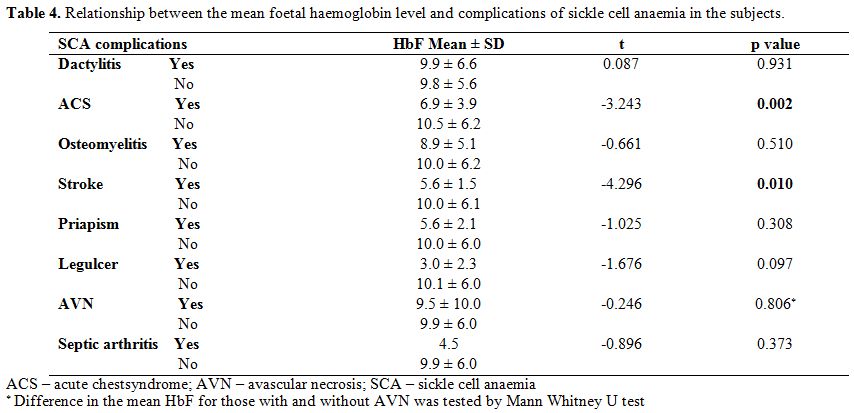 |
Table 4.
Relationship between the mean foetal haemoglobin level and complications of sickle cell anaemia in the subjects. |
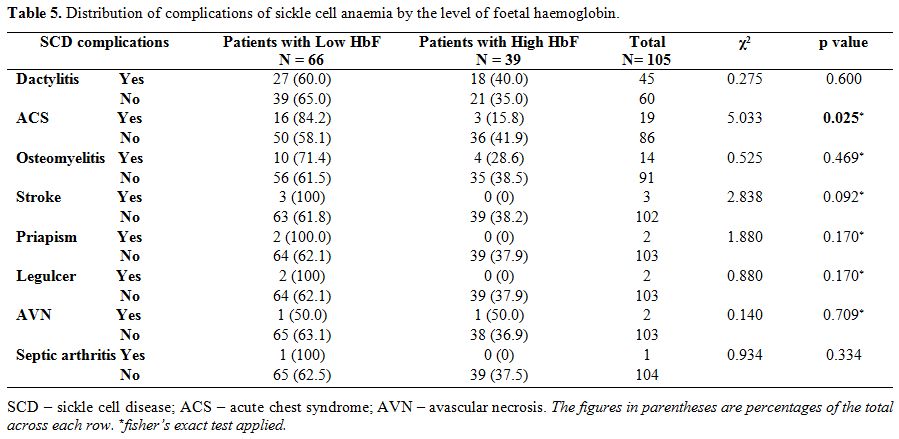 |
Table 5. Distribution of complications of sickle cell anaemia by the level of foetal haemoglobin. |
Multivariate logistic regression analysis.
Logistic regression analysis was done to examine the independent effect
of foetal haemoglobin on SCD severity. In the regression model, disease
severity (dichotomised as mild or moderate) was taken as the
outcome/dependent variable, and age group (1-5 vs>5 years), gender
(male vs female), socio-economic class (high vs middle/low) and foetal
haemoglobin (high vs low HbF) were all taken as the
predictive/independent factors. None of these factors [age group, OR
0.3, 95% CI 0.1-1.0, p=0.050; gender, OR 1.9, 95% CI 0.7-5.0, p=0.198;
social class, OR 0.9, 95% CI 0.2-4.4, p=0.947 and foetal haemoglobin,
OR 1.8, 95% CI 0.6–5.2, p=0.262] was found to be a significant
independent predictor of SCD severity.
Discussion
Interest
in foetal haemoglobin among patients with SCA has been on the increase
in the last six decades during which its substantial protective effects
on the timing and severity of the disease symptomatology and the
development of multi-organ dysfunction became subjects of scientific
research.[11] However, in Nigeria, and indeed in most
parts of sub-Saharan Africa, where the burden of the disease is
highest, studies on foetal haemoglobin and its role on clinical
manifestations and disease severity in children are scanty.
The
clinical burden of SCA in our unit reflects the high burden of this
disease in most parts of developing countries. In this study, about 85
percent of the children had experienced at least one significant pain
episode necessitating hospital visit and use of analgesia; while about
20 percent were transfused and 50 percent were admitted for SCD-related
morbidities in the twelve months preceding the study. These findings
agree with the report by Brown et al[12] among SCA
children at the University College Hospital, Ibadan. It was also found
in this study that about one-third of the subjects had more than three
episodes of vaso-occlusive crisis and indeed this was responsible for
most of the hospital admissions among them. This situation is
consistent with the trend in some countries such as Britain and Saudi
Arabia.[13,14] Although the frequency of pain and
admissions have been reduced significantly in developed nations
following the widespread use of hydroxyurea, this drug is not readily
available, accessible and affordable in Nigerian and other sub-Saharan
African countries where the principal precipitants of vaso-occlusive
crises such as malaria and sepsis are still prevalent.[15]
The
mean HbF level of the children with SCA in this study is higher than
what has been reported previously in most Nigerian studies. Isah[16]
in Sokoto found mean HbF level of 2.99±5.16 percent as against 9.9±6.0
percent in the present study. Also, the mean HbF level in this study is
higher than 7.2±5.0 percent reported by Tshilolo in Congo.[17]
This discrepancy may be due to the difference in the method of foetal
haemoglobin estimation. The Betke method of alkali denaturation was
used by Isah while HPLC which is a more sensitive method was used in
the present study. Age difference might also account for the
differences in the haemoglobin F level. For instance, relatively lower
values were obtained in previous studies of adult sickle cell patients
by Omoti in Benin (2.17±1.81 percent), Durosinmi in Ife (4.26±4.33
percent), Olaniyi in Ibadan (5.16±4.04 percent) and Uko in Calabar
(3.05±1.61 percent).[18-21]
A study among children with SCA in Uganda reported a mean HbF level of 9.0±5.58 percent despite using alkali denaturation test.[9] A higher value (12.2±7.1 percent) was also reported in India by Rao et al.[22]
The reason for the differences may be due to the effect of various
factors that influence foetal haemoglobin production in SCA
individuals. One of the factors is beta gene haplotype of the disease.
The Senegal, Saudi and Indian haplotypes are generally associated with
higher levels of foetal haemoglobin and milder disease course while the
Benin haplotype which is found commonly in our environment and the
Cameroon haplotype are intermediate and of varying clinical
manifestation. However, the Bantu haplotype is associated with severe
disease and low foetal haemoglobin production.[23]
Another major factor that affects foetal haemoglobin level includes
hydroxyurea, a ribonucleotide reductase inhibitor. The exact mechanism
of how hydroxyurea increases HbF levels is not clear. It is frequently
used in the western world in the management of children with sickle
cell disease. Unfortunately, most children in Nigeria and other
resource-poor countries in sub-Saharan Africa with the largest burden
of the disease are not benefitting due to non-availability and/or
unaffordability of the drug.
A recent in-vitro study highlighted a beneficial effect of Tropical almond (Terminalia cattapa) in inducing HbF levels in erythroid progenitor cells.[24]
Possibly, a large-scale study and clinical trials on in-vivo use of
this agent in children with SCA living in resource-poor countries may
determine if it could be of comparative clinical utility.
Currently, a Nigerian child who is using a 500mg capsule of hydroxyurea
daily would spend close to 10 US dollars per month on HU therapy alone,
in a country where more than 70 percent of the population is poor.
This
present study found about two-thirds of our children with SCA (62.9
percent) had low foetal haemoglobin levels, and this was demonstrated
in the pattern of the burden and complications seen in this study. We
also observed that a significantly higher proportion of children with
moderate disease severity compared to those with mild disease severity
(78.1% vs 56.2%) had low levels of HbF. On the other hand, more of the
children with mild SCD severity than those with moderate disease had
high levels of HbF. A similar finding was reported by Mpalampa et al.[9]
among children with SCA in Uganda. Also, females in the present study
had significantly higher HbF compared to males. This datum is similar
to that found by Falusi et al.[25] in a study of adult patients with SCA as well as in other studies by Mouele[26] in Congo and Alsultan[27]
in Saudi Arabia. Olaniyi et al. in Ibadan, Nigeria found no difference
in the HbF levels in both adult males and females with SCA.[18] Falusi et al.[25]
attributed the finding to hormonal factor at puberty. However, in
children, the exact reason for the higher levels of haemoglobin F among
girls may not be explained by hormonal changes alone. The exact reason
remains unclear, perhaps the X-linked co-dominant gene controlling the
production of HbF may lead to a double dose of the gene as is the case
for females unlike males, resulting in higher elaboration of the gene
products, hence, higher HbF levels in females than males.[28]
The mean HbF was also found to be inversely related to age and
significantly reduces with increasing age. This datum is similar to the
finding of Adekile et al.[29] among SCA individuals
in Kuwait. It may be due to the relatively higher amount of F cells in
the younger age group as observed by Akinsheye et al.[11]
We
found that the mean foetal haemoglobin levels were significantly lower
in children with moderate disease severity than those with mild disease
and also in those who had a history of acute chest syndrome and stroke
than those without these complications. Although there are no local
data to corroborate or refute this observation, our findings are
similar to the report by Mpalampa from Uganda.[9]
Increased polymerisation of the sickle cell haemoglobin in the presence
of lower foetal haemoglobin may account for these unfavourable events
among children with SCA.[30] We did not find any
relationship between foetal haemoglobin levels and previous history of
priapism, osteomyelitis, septic arthritis and leg ulcer. Although a
higher level of foetal haemoglobin has been found to be associated with
fewer complications, the non-significant relationship between foetal
haemoglobin and these other complications in this present study may be
due to the fewer number of study participants with these complications.
Our study has some limitations. First, it is a cross-sectional
study from a single centre. This type of study design is subject to
recall, investigator and survival biases. In particular, information on
the lifetime incidence of complications and frequency of significant
pain episodes may have been subject to recall bias, despite the review
of relevant medical charts in addition to clinical histories to obtain
that information. Secondly, the relatively small number of patients
studied could also affect generalisability of our findings. Future
studies should be multi-centred, longitudinal in design and involve a
larger population.
It is concluded that foetal haemoglobin level
has a significant inverse relationship with disease burden and severity
among children with sickle cell anaemia. Advanced age and male gender
were also significantly related to low foetal haemoglobin levels in
these children. Therefore, it is recommended that facilities for early
and regular quantification of foetal haemoglobin be made available, and
access to HbF inducing agents, specifically hydroxyurea encouragedin
order to reduce the morbidity and mortality among these children.
Acknowledgement
We appreciate the cooperation of the parents and children who participated in this study.
References
- Modell B, Darlison M. Global epidemiology of
haemoglobin disorders and derived service indicators. Bull World Health
Organ. 2008;86(6):480-7. https://doi.org/10.2471/BLT.06.036673 PMid:18568278 PMCid:PMC2647473

- Sickle-cell anaemia. Agenda item 11.4. In: 59th World Health Assembly; 2006; Geneva: World Health Organization. http://www.who.int/iris/handle/10665/20890
- Wang
WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al.
Hydroxycarbamide in very young children with sickle-cell anaemia: A
multicentre, randomised, controlled trial (BABY HUG). Lancet
2011;377(9778):1663–72. https://doi.org/10.1016/S0140-6736(11)60355-3
- Adegoke
SA, Adeodu OO, Adekile AD. Sickle cell disease clinical phenotypes in
children from South-Western, Nigeria. Niger J Clin Pract.
2015;18(1):95-101. PMid:25511352
- Serjeant GR. Fetal hemoglobin in homozygous sickle cell disease Clin Haematol 1975;4:109-22. PMid:1102178
- Oyedeji GA. Socioeconomic and cultural background of hospitalised children in Ilesa. Niger J Paediatr. 1985;12:111-7.
- Ballas
SK, Lieff S, Benjamin LJ, Dampier CD, Heeney MM, Hoppe C, et al.
Definitions of the phenotypic manifestations of sickle cell disease. Am
J Hematol 2010;85:6-13. PMid:19902523 PMCid:PMC5046828
- Adegoke
SA, Kuti BP. Evaluation of clinical severity of sickle cell anemia in
Nigerian children. J Applied Hematol. 2013;4:58-64.
- Mpalampa
L, Ndugwa CM, Ddungu H, Idro R. Foetal haemoglobin and disease severity
in sickle cell anaemia patients in Kampala, Uganda. BMC Blood
disorders. 2012;12:11. https://doi.org/10.1186/1471-2326-12-11 PMid:22958547 PMCid:PMC3520739
- Norman NH, Nie, Hull CH. Statistical package for social sciences for Windows. 17.0 ed. Chicago: SPSS Incoporation; 2008.
- Akinsheye
I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastian P, et al.
Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19-27. https://doi.org/10.1182/blood-2011-03-325258 PMid:21490337 PMCid:PMC3139383
- Brown
B, Jacob N, Lagunju I, Jarrett O. Morbidity and mortality pattern in
hospitalized children with sickle cell disorders at the University
College Hospital, Ibadan, Nigeria. Niger J Paed. 2013;40(1):34-9.
- Brozovic
M, Davies S, Alison I, Brownell A. Acute admissions of patients with
sickle cell disease who live in Britain. Br Med J. 1987;294:1206-8. https://doi.org/10.1136/bmj.294.6581.1206
- Wasil J. Epidemiology of sickle cell disease in Saudi Arabia. Ann Saudi Med. 2011 31:289-93. https://doi.org/10.4103/0256-4947.81540 PMid:21623060 PMCid:PMC3119971
- Adewoyin
AS. Management of Sickle Cell Disease: a review for physician education
in Nigeria (Sub-Saharan Africa). Anemia. 2015;2015:e791498.
- Isah
IZ, Udomah FP, Erhabor O, Aghedo F, Uko EK, Okwesili AN, et al. Foetal
haemoglobin levels in sickle cell disease patients in Sokoto, Nigeria.
Br J Med Health Sci. 2013;1:36-47.
- Tshilolo
L, Summa V, Gregorj C, Kinsiama C, Bazeboso JA, Avvisati G, et al.
Foetal haemoglobin, erythrocytes containing foetal haemoglobin, and
hematological features in Congolese patients with sickle cell anaemia.
Anemia. 2012;2012:e105349
- Olaniyi
JA, Arinola OG, Odetunde AB. Foetal haemoglobin (HbF) status in adult
sickle cell anaemia patients in Ibadan, Nigeria. Ann Ibadan Postgrad
Med. 2010;8:30-3. PMid:25161472 PMCid:PMC4138770
- Durosimi
MA, Salawu L, Ova YAA, Lawal OO, Fadiran OA. Haematological parameters
in sickle cell anaemia patients with and without splenomegaly. Niger
Postgrad Med J. 2005;12(4):271-4.
- Uko
E, Useh M, Gwanmesia F. Frequency of foetal haemoglobin and haemoglobin
values in various haemoglobin genotypes in Calabar, Nigeria. East Afr
Med J. 1997;74(12):809-11. PMid:9557428
- Omoti
CE. The value of foetal haemoglobin level in the management of Nigerian
sickle cell anaemia patients. Niger Postgrad Med J. 2005;12(3):149-54.
PMid:16160713
- Rao
SS, Goyal JP, Raghunath SV, Shah VB. Hematological profile of sickle
cell disease from South Gujarat, India. Hematol Reports. 2012;4:e8. https://doi.org/10.4081/hr.2012.e8 PMid:22826798 PMCid:PMC3401137
- Nagel
RL, Fabry ME, Pagnier J, Zohoun I, Wajcman H, Baudin V, et al.
Hematologically and genetically distinct forms of sickle cell anemia in
Africa. N Engl J Med 1985 321:880. https://doi.org/10.1056/NEJM198504043121403 PMid:2579336
- Aimola
IA, Inuwa HM, Nok AJ, Mamman AI. Induction of foetal haemoglobin
synthesis in erythroid progenitor stem cells: mediated by water-soluble
components of Terminalia catappa. Cell Biochem Funct. 2014;32(4):361-7.
https://doi.org/10.1002/cbf.3024 PMid:24470326
- Falusi AG, Esan GJ. Foetal haemoglobin levels in sickle cell anaemia in Nigerians. Afr J Med Sci. 1989;18:145-9.
- Mouele
R. Haemoglobin F (HbF) levels in sickle-cell anaemia patients
homozygous for the Bantu haplotype. European Journal of Haematology.
1999;63:136–7. https://doi.org/10.1111/j.1600-0609.1999.tb01128.x PMid:10480294
- Alsultan
A, Solovieff N, Aleem A, AlGahtani FH, Al-Shehri A, Osman ME, et al.
Fetal hemoglobin in sickle cell anemia: Saudi patients from the
Southwestern province have similar HBB haplotypes but higher HbF levels
than African Americans. Am J Hematol. 2011;86(7):612-4. https://doi.org/10.1002/ajh.22032 PMid:21630302
- Dover
GJ, Smith KD, Chang YC, Purvis S, Mays A, Meyers DA, et al. Fetal
Hemoglobin Levels in Sickle Cell Disease and Normal Individuals Are
Partially Controlled by an X-Linked Gene Located at Xp22.2. Blood
1993;80(3):816-24.
- Adekile
A, Al-Kandari M, Haider M, Rajaa M, D'Souza M, Sukumaran J. Hemoglobin
F concentration as a function of age in Kuwaiti sickle cell disease
patients. Med Princ Pract. 2007;16(4):286-90. https://doi.org/10.1159/000102151 PMid:17541294
- Trompeter S, Roberts I. Haemoglobin F modulation in childhood sickle cell disease. Br J Haematol. 2008;144:308–16. https://doi.org/10.1111/j.1365-2141.2008.07482.x PMid:19036119
[TOP]