Paolo Milani, Giampaolo Merlini and Giovanni Palladini.
Amyloidosis
Research and Treatment Center, Foundation “Istituto di Ricovero e Cura
a Carattere Scientifico (IRCCS) Policlinico San Matteo” and Department
of Molecular Medicine, University of Pavia, Pavia, Italy.
Corresponding
author: Prof. Giovanni Palladini, MD,
PhD. Amyloidosis Research and Treatment Center, Foundation “IRCCS
Policlinico San Matteo”, Viale Golgi, 19, 27100 Pavia, Italy. Tel.:
+39-0382-502994, fax: +39-0382-502995. E-mail:
giovanni.palladini@unipv.it
Published: March 1, 2018
Received: January 10, 2018
Accepted: February 5, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018022 DOI
10.4084/MJHID.2018.022
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Light
chain (AL) amyloidosis is caused by a usually small plasma-cell clone
that is able to produce the amyloidogenic light chains. They are able
to misfold and aggregate, deposit in tissues in the form of amyloid
fibrils and lead to irreversible organ dysfunction and eventually death
if treatment is late or ineffective. Cardiac damage is the most
important prognostic determinant. The risk of dialysis is predicted by
the severity of renal involvement, defined by the baseline proteinuria
and glomerular filtration rate, and by the response to therapy. The
specific treatment is chemotherapy targeting the underlying plasma-cell
clone. It needs to be risk-adapted, according to the severity of
cardiac and/or multi-organ involvement. Autologous stem cell transplant
(preceded by induction and/or followed by consolidation with
bortezomib-based regimens) can be considered for low-risk patients
(20%). Bortezomib combined with alkylators is used in the majority of
intermediate-risk patients, and with possible dose escalation in
high-risk subjects. Novel, powerful anti-plasma cell agents were
investigated in the relapsed/refractory setting, and are being moved to
upfront therapy in clinical trials. In addition, the use of novel
approaches based on antibodies targeting the amyloid deposits or small
molecules interfering with the amyloidogenic process gave promising
results in preliminary studies. Some of them are under evaluation in
controlled trials. These molecules will probably add powerful
complements to standard chemotherapy. The understanding of the specific
molecular mechanisms of cardiac damage and the characteristics of the
amyloidogenic clone are unveiling novel potential treatment approaches,
moving towards a cure for this dreadful disease.
|
Introduction
Immunoglobulin
light chain (AL) amyloidosis is the most common form of systemic
amyloidosis, accounting for approximately 70% of all subjects suffering
from these diseases.[1] It is caused by a plasma cell clone that
infiltrates the bone marrow by less than 10% in half of the patients.
Despite its relatively small size, the clone can set off a devastating
multi-organ damage caused by the monoclonal light chain.[2] The
amyloidogenic light chain misfolds and aggregates, depositing in
tissues in the form of amyloid fibrils.[3] All organs, except for the
central nervous system, can be affected by this process, that leads to
irreversible organ dysfunction and death if unrecognized or treated
ineffectively.[3] In the last 15 years, we have made substantial
progress in understanding the biology of the amyloid plasma cell clone
and the mechanisms of organ damage. Moreover, with accurate prognostic
stratification and response assessment based on biomarkers of clonal
and organ disease, we have learnt to safely apply treatments originally
developed for multiple myeloma, to the fragile patients with AL
amyloidosis.[2,4,5] Nevertheless, the timely recognition and the
appropriate treatment of patients with AL amyloidosis remains
challenging even for hematologists who are expert in multiple myeloma.
In this review, we summarize the current knowledge on the pathogenesis
of AL amyloidosis, and we focus on the clinical management of patients
with this disease.
The Amyloid Clone and Mechanisms of Organ Damage
Not
only is the amyloidogenic clone usually smaller in size than that
causing multiple myeloma, but it is characterized by a significant
frequency of chromosomal abnormalities, that can affect treatment
outcomes. The most frequent is t(11;14) translocation, observed in
almost 50% of patients.[6] The presence of t(11;14) is associated with
poorer outcome with bortezomib-based and immunomodulatory (IMiDs)-based
therapy, even when cyclophosphamide is added.[7,8] The adverse impact of
t(11;14) can be overcome by melphalan, administered orally or in
autologous stem cell transplant.[9,10] Gain of 1q21 is less frequent in
AL amyloidosis than in multiple myeloma, being found in less than 20%
of patients.[6] Patients harboring this abnormality have poorer outcome
when treated with oral melphalan/dexamethasone (MDex) without the
addition of bortezomib.[11] Clonal plasma cells in AL amyloidosis have
similar phenotypic and copy number alteration profiles as those found
in multiple myeloma, but their gene expression profile is similar to
that of normal plasma cells.[12] A genome-wide association study showed a
shared genetic susceptibility between AL amyloidosis and multiple
myeloma, but cyclin D1 was a more prominent driver in AL amyloidosis.[13]
The plasma cells rely on the proteasome to cope with the proteotoxicity
exerted by the misfolded, amyloidogenic light chains.[14-16] This makes
the amyloid plasma cell clone keenly sensitive to proteasome inhibitors.
The
light chain variable region gene and the gene family of the clone can,
at least in part, explain the variable organ tropism of AL amyloidosis.
Indeed, three Vλ genes, IGLV2–14, IGVL6-57, and IGLV3-1 contribute to
encoding the majority of amyloidogenic λ
light chains.[17-19] The germline gene LV6-57 is common in AL amyloidosis
while it is exceedingly rare in normal B-cells, and it is associated
with renal involvement.[20] Usage of LV1-44 germline gene is linked to
predominant cardiac involvement, whereas KV1-33 is associated with
involvement of the liver.[21]
Since cardiac involvement is the
main determinant of survival, efforts have been focused on unveiling
the mechanisms of cardiac dysfunction in AL amyloidosis. The
observation of complete clinical recovery of patients after effective
chemotherapy in the absence of significant reduction of amyloid
deposits indicates that the mass action caused by the deposits is not
the only, and possibly not the main, determinant of organ dysfunction
in AL amyloidosis. The availability of cardiac biomarkers, particularly
N-terminal pro-natriuretic peptide type B (NT-proBNP) as a measure of
amyloid cardiac dysfunction, showed that the clinical severity of heart
failure and patient survival is linked to changes in the concentration
of the circulating amyloidogenic free light chains rather than to
changes in the amyloid load.[22-24] Indeed, the infusion of light chain
purified from the urine of patients with cardiac amyloidosis causes a
rapid increase in end-diastolic pressure in isolated mouse hearts in a
matter of few minutes, which is not observed with control light
chains.[25] Exposing Caenorhabditis elegans, a worm whose pharynx
pulses rhythmically and is considered an analog of the vertebrate
heart, to light chains of patients with cardiac AL amyloidosis, but not
to control light chains, reduces the rate of pharynx contraction.[26]
Finally,
the injection of light chains from patients with cardiac AL amyloidosis
in the heart of zebrafish reduces the cardiac output and the lifespan
of the fishes in the absence of amyloid deposits, which is not observed
with control light chains.[27] Overall, this clinical and experimental
evidence point to the toxicity exerted by the circulating precursor as
the main cause of cardiac dysfunction in AL amyloidosis.[28,29]
Clinical Presentation and Diagnosis
The clinical manifestations of AL amyloidosis depend on organ involvement (Figure 1)
but are rarely specific. Involvement of the soft tissues with
macroglossia, periorbital purpura, submandibular gland swelling, and
shoulder pad sign can easily trigger the diagnosis but are uncommon.
More frequently, AL amyloidosis manifests with sign and symptoms
resembling those of more common conditions of the elderly. Cardiac
involvement (approximately 80% of patients) manifests with heart
failure with preserved ejection fraction. Echocardiography is the
cornerstone of the assessment of amyloid cardiomyopathy revealing
increased ventricular wall thickness and granular sparkling. While
ejection fraction is usually preserved until late stages of the
disease, longitudinal strain, and midwall fractional shortening are
often altered and have prognostic relevance.[30,31]
Electrocardiogram usually shows low limb voltages in cardiac AL
amyloidosis. Late gadolinium enhancement at cardiac magnetic resonance
strongly points to the diagnosis of heart involvement; moreover,
cardiac magnetic resonance can quantify the extracellular volume that
may reflect the amyloid load.[32] The scintigraphy
tracers developed for imaging the amyloid deposits in the brain of
patients with Alzheimer disease, can identify cardiac amyloidosis and
are promising tools for detecting and possibly quantitating amyloid
deposits also in systemic amyloidoses.[33] The uptake
of bone tracers in patients with AL amyloidosis is absent or moderate,
differently from transthyretin cardiac amyloidosis, characterized by a
strong uptake. This difference can be used to distinguish between the
two forms.[34] Increased concentrations of NT-proBNP
are found in 100% of patients with cardiac AL amyloidosis, and precede
symptoms and imaging alterations, allowing diagnosis at very early
stages.[22,35] The kidney is
involved in two-thirds of patients with AL amyloidosis. The disease
manifests with albuminuria, evolving in nephrotic syndrome and
progressing to renal failure eventually leading to end-stage renal
disease if unrecognized or ineffectively treated. Involvement of the
liver is characterized by organ enlargement without scan defects and
elevation of alkaline phosphatase. Peripheral neuropathy is axonal,
predominantly sensory and centripetal. Involvement of the autonomic
nervous system is common but usually asymptomatic, although it can
often become manifest with inappropriate use of hypotensive drugs. It
is characterized by postural hypotension that can be preceded by the
"resolution" of a pre-existing hypertension, erection defects in males
and disturbances in bowel movements. General symptoms, most commonly
profound fatigue and malnutrition, often accompany more organ-specific
manifestations.
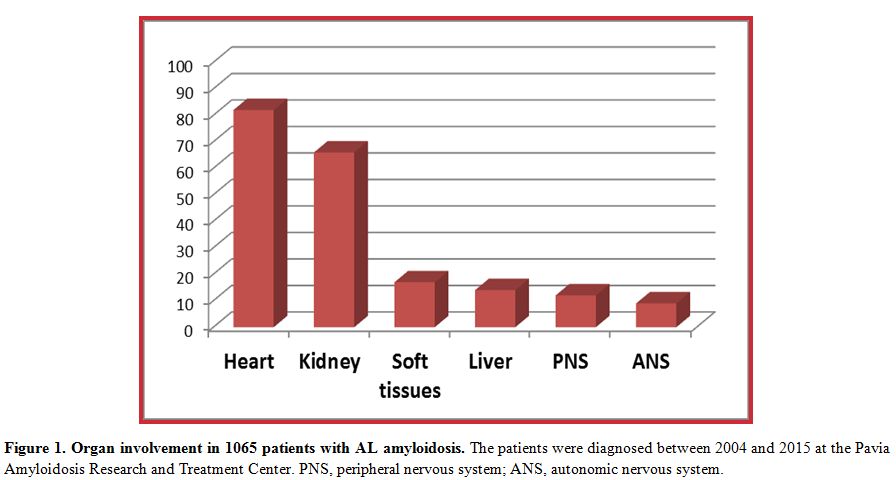 |
Figure 1. Common types of systemic amyloidosis. |
These
clinical manifestations are not only resembling those of more common
conditions, but they are usually associated with advanced stages of the
disease. All this, unfortunately, results in frequent diagnostic
delays. A recent survey showed that 40% of patients with AL amyloidosis
remain undiagnosed 1 year after the onset of symptoms.[36]
Similar delays are also observed in patients who are performing regular
follow-up for monoclonal gammopathy of undetermined significance (MGUS)
under the supervision of hematologists.[37] This is
because the classical workup of patients with MGUS does not include
appropriate, sensitive tools for the detection of the onset of organ
involvement. Thus, we advocated the inclusion of sensitive markers of
cardiac (NT-proBNP) and renal (albuminuria) amyloidosis in the regular
follow-up of patients with MGUS and abnormal free light chain (FLC)
ratio.[35,38]
Once amyloidosis
is suspected, the diagnosis requires the demonstration of amyloid
deposits in a biopsy. The abdominal fat aspirate is simple and
minimally invasive, although its interpretation requires expertise. In
combination with biopsy of the bone marrow stained with Congo red
and/or biopsy of a minor salivary gland, it can yield a diagnostic
sensitivity of approximately 90%, thus sparing organ biopsies.[39-41]
Nevertheless, organ biopsies may need to be performed in subjects with
strong clinical suspicion and negative fat, gland, and bone marrow.
Typing of the amyloid deposits is mandatory, in order to avoid
misdiagnosis between AL amyloidosis and other common forms of systemic
amyloidosis (listed in Table 1),
such as hereditary or wild-type (formerly senile) transthyretin
amyloidosis, hereditary apolipoprotein AI amyloidosis, leucocyte
chemotactic factor-2 amyloidosis, and amyloidosis reactive to chronic
inflammation. Incorrect typing could lead to disastrous therapeutic
errors.[42-44] Unfortunately, light microscopy
immunohistochemistry and immunofluorescence with commercial antibodies,
the most commonly available techniques, are unreliable to characterize
amyloid deposits.[45,46] Thus, in most instances a
reliable diagnosis requires referral of patients to specialized
centers. Light microscopy immunohistochemistry can be reliably
performed at referral centers using custom-made antibodies.[47] Immunoelectron microscopy with commercial antibodies can correctly identify the amyloid type in almost 100% of patients.[40]
Mass spectrometry-based proteomics can be used on whole tissues or
after laser capture microdissection to reliably type amyloid deposits.[48,49]
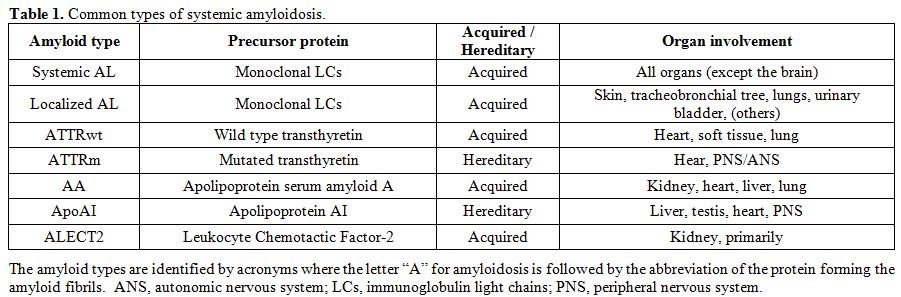 |
Table 1. Common types of systemic amyloidosis. |
Once
the diagnosis and typing of AL amyloidosis have been established, the
diagnostic workup is completed by assessing the burden and severity of
clonal and organ disease, as summarized in Table 2.
Given the small size of the amyloid plasma cell clone, the combination
immunofixation of both serum and urine with measurement of circulating
free light chain is required to grant adequate sensitivity.[50-53] Assessment of organ involvement is based on biomarkers, electrocardiogram, and imaging studies.
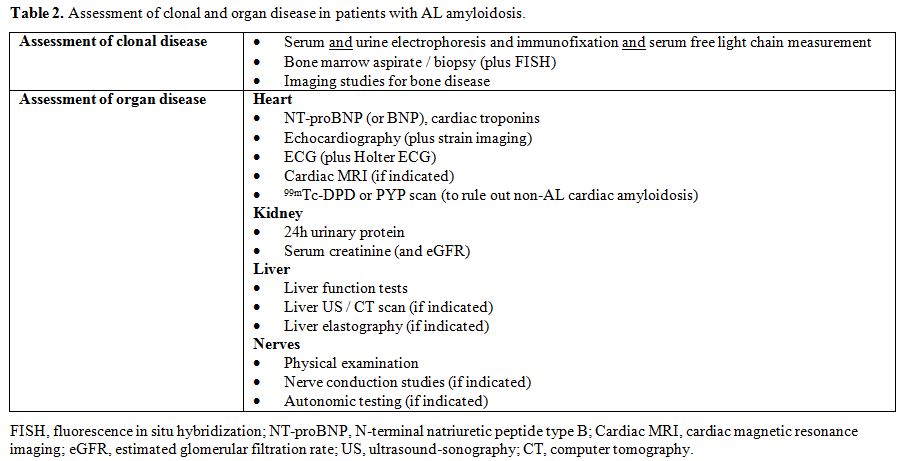 |
Table 2. Assessment of clonal and organ disease in patients with AL amyloidosis. |
Staging
The
survival of patients with AL amyloidosis is exceedingly heterogeneous,
depending on the severity of cardiac dysfunction at the time of
diagnosis: while patients who are diagnosed late, at a stage when heart
damage is very advanced and not amenable of improvement with treatment
survive only a few weeks, patients without heart involvement can
survive years even if they fail to respond to therapy. This extreme
heterogeneity requires accurate prognostic stratification for
establishing the best therapeutic approach, balancing treatment
intensity and rapidity of action with patients’ frailty, as well as for
comparing results of clinical trials. The Mayo Clinic group established
a simple and reliable staging system based on NT-proBNP and cardiac
troponins, which was then modified by European investigators (Table 3).[54,55]
This staging system is now the most widely used for clinical trial
design and patient management. Besides heart involvement, clonal
burden, assessed by bone marrow plasma cell (BMPC) infiltration or dFLC
(difference between involved and uninvolved circulating free light
chains) has an independent impact on survival. Patients with AL
amyloidosis and BMPC infiltrate >10% have a more reduced survival,
which is comparable to that of patients who have concomitant overt
multiple myeloma.[56] Subjects who have a very low (<50 mg/L) dFLC level have a significantly better outcome across cardiac stages.[57,58] The Mayo Clinic group has incorporated the dFLC level in the cardiac staging system (Table 3).[59]
The severity of renal involvement does not directly affect patient’s
survival, but impacts the quality of life and reduces the access to
effective therapy. A staging system predicting progression to dialysis
has been proposed and validated by European investigators (Table 3).[60]
Similarly to heart involvement, recognition and prompt treatment of
renal AL amyloidosis at early stages can almost abolish the risk of
progression to dialysis, while late diagnosis at advanced stages is
associated with higher risk of progression.
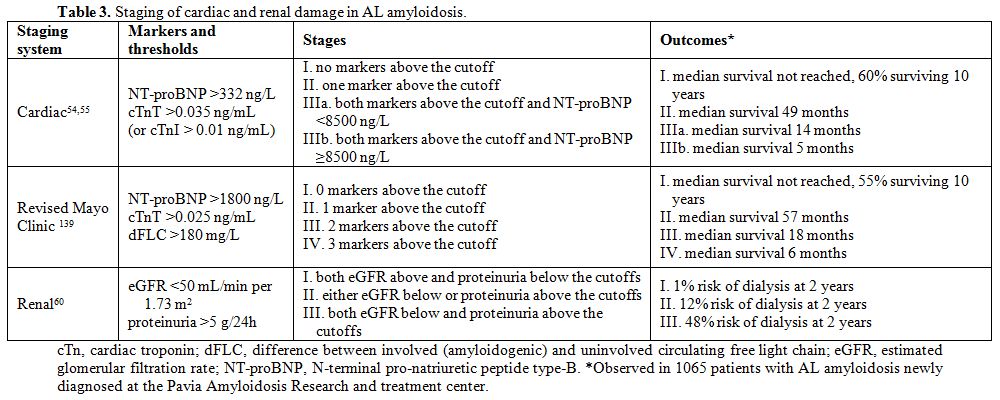 |
Table 3. Staging of cardiac and renal damage in AL amyloidosis |
Treatment
The
complexity of AL amyloidosis, which is due to the unique coexistence of
a clonal plasma cell disorder and dysfunction multiple vital organs,
makes treatment of this disease a challenge even for hematologists who
are experts in the field of multiple myeloma. Indeed, the availability
of new drugs, directly targeting the amyloid deposits, will probably
displace AL amyloidosis from the realm of exclusive hematologic
therapy. The experience of treating physicians significantly impacts
patients’ outcomes,[61] and very few recent
prospective controlled studies exist to guide the therapeutic strategy.
Thus, whenever possible, patients should be referred to specialized
centers for treatment. Indeed, the amyloid clone requires treatment
even if in the vast majority of cases it does not meet the criteria for
treating multiple myeloma.[62] Moreover, differently
from patients with multiple myeloma, subjects suffering from AL
amyloidosis are at high risk of death and are extremely susceptible to
treatment toxicity in the first few months after diagnosis; whereas, if
they survive this first dangerous time, they enjoy a better long-term
outcome compared to myeloma patients.[63] For this
reason, chemotherapy is usually delivered at the lowest effective dose
during the first cycles. The treatment strategy needs to be adapted to
early treatment efficacy and should not be planned in advance. The
response should frequently be assessed, at least every 2 cycles, in
order to allow rapid switch to rescue therapy in patients who do not
achieve satisfactory response. The criteria for hematologic,[57,58,64] cardiac,[64] and renal[60] response (summarized in Table 4)
have been established and validated in a huge international effort and
offer guidance to individual patients treatment, as well as surrogate
endpoints for clinical trials.[65] In particular, a
new criterion where both hematologic and organ response can be assessed
simultaneously early on in the treatment of AL amyloidosis was proposed
to stratify the risk of patients, supporting its use as a surrogate
end-point in clinical trials.[66] In addition, a
recent report from Mayo Clinic showed that the better survival was
assessed in patients who obtained the deeper organs (heart, kidney,
liver).[67]
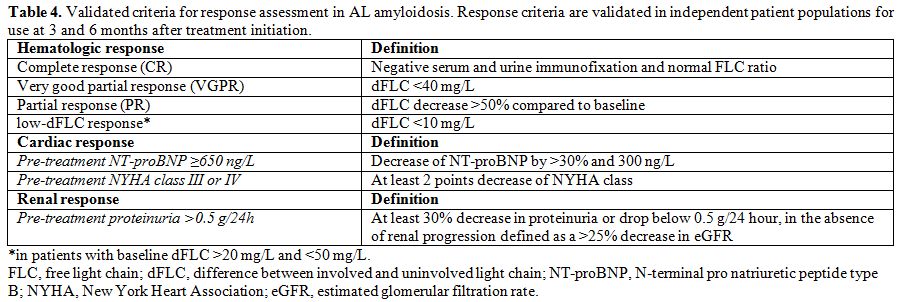 |
Table 4. Validated criteria for response
assessment in AL amyloidosis. Response criteria are validated in
independent patient populations for use at 3 and 6 months after
treatment initiation. |
Chemotherapy targeting the amyloid plasma cell clone.
Anti-plasma cell chemotherapy is the cornerstone of treatment of AL
amyloidosis and was able to remarkably improve patients’ outcomes over
the last decades.[5] Treatment of AL amyloidosis should be adapted to the severity of organ involvement.
Low-risk
patients represent approximately 15% of all subjects suffering from AL
amyloidosis and can be considered for autologous stem cell
transplantation (ASCT). This procedure is associated with a
substantially higher risk of early mortality compared to multiple
myeloma. However, refinement in selection criteria has reduced
transplant-related mortality over time.[61]
Accumulation of expertise is also crucial, the outcome being
significantly poorer at centers where less than four transplants per
year are performed in patients with this disease.[61]
When an adequate selection of transplant candidates is applied at
referral centers, the outcome is excellent, with hematologic response
in 71% of subjects and complete response (CR) in 35-37%.[61,68] These results in overall median survival of 7.6 years.[68]
The great majority of transplant-related mortality occur in patients
with elevated cardiac biomarkers, and subjects whose NT-proBNP is
>5000 ng/L and/or cTnT is >0.06 ng/mL should not be offered ASCT.[69,70]
Other eligibility criteria are ejection fraction >45% at
echocardiography, New York Heart Association (NYHA) class <III,
orthostatic systolic blood pressure >90 mmHg, age <65 years,
performance status (Eastern Cooperative Oncology Group) ≤2, eGFR >50
mL/min per 1.73 m2 unless on dialysis, and lung CO diffusion capacity >50%.[1,70,71]
Patients who do not obtain CR after ASCT can receive bortezomib-based
treatment. Overall, this sequential approach yields a 60% rate of CR.[72]
Induction therapy with bortezomib before ASCT improves outcomes in
patients with a bone marrow plasma cell infiltrate >10%.[73]
Intermediate
risk patients account for approximately 70% of patients with AL
amyloidosis. They receive non-transplant chemotherapy. Until recently,
a standard treatment for these patients has been oral melphalan and
dexamethasone (MDex).[74,75] This regimen is very well tolerated and yields a 76% overall hematologic response rate, with CR in 31% of cases.[76]
A randomized trial compared MDex to ASCT and failed to demonstrate an
advantage for ASCT in terms of response rate and survival.[77]
This trial was performed before the availability of a biomarker-based
selection of transplant candidates, and the results were considered
influenced by very high transplant-related mortality. Nevertheless, a
landmark analysis excluding early deaths confirmed these results.
Bortezomib-based regimens are now considered upfront standards of care
in most patients with AL amyloidosis. A large retrospective study and a
prospective trial showed efficacy of bortezomib in relapsed /
refractory patients.[78-81] In the largest study of
frontline treatment with cyclophosphamide, bortezomib and dexamethasone
(CyBorD), the overall hematologic response rate was 60%, with CR in 23%
of cases.[82] Two retrospective case-control studies
showed higher response rates with bortezomib in combination with
alkylating agents and dexamethasone compared to the previous standards
of care MDex and cyclophosphamide / thalidomide / dexamethasone, though
without a survival benefit.[83,84] An international
phase III study (NCT01277016) comparing MDex with bortezomib plus MDex
(BMDex) has recently been completed, showing significantly higher
overall hematologic response rate with BMDex (81% vs. 57%, P=0.005).[85]
Based on this data, bortezomib should be offered to intermediate-risk
patients, in the absence of contraindications such as peripheral
neuropathy. The choice of the best combination should take into account
clonal and patient characteristics. A recent study by Kastritis, et al.
showed that addition of cyclophosphamide and higher doses of
dexamethasone do not improve outcomes of patients with AL amyloidosis
treated with bortezomib.[86] Treatment with BMDex has
the advantage of overcoming the effects of both gain 1q21 (poor outcome
with oral melphalan) and t(11;14) (poor outcome with bortezomib).[7,8,10,11]
Oral melphalan should not reach the cumulative dose of 150 mg (not
exceeding 2 cycles) in patients who may be selected for subsequent stem
cell mobilization and harvest.[87] Treatment with
bortezomib / dexamethasone alone or in combination with
cyclophosphamide is preferred in patients with potentially reversible
contraindication to ASCT, being stem cell sparing, as well as in
subjects with renal failure.
The remaining 15-20% of patients
with AL amyloidosis are high-risk, most frequently because of advanced
cardiac stage (IIIb) or severe heart failure (NYHA class III or IV). So
far, no treatment approach, including those based on bortezomib, was
able to overcome the poor prognosis of these patients, and median
survival ranges from 3 to 7 months.[88] Nevertheless,
the few patients who survive long enough (at least 3 months) to take
advantage of response to chemotherapy enjoy prolonged survival.[82]
A recent report from The United Kingdom Group showed that patients
achieving a rapid response at day 30 or overall CR/VGPR at 6 months had
markedly better survival.[89] High-risk patients are
treated with low-dose combinations, with weekly dose escalation based
on tolerability under intensive monitoring.[1]
Relapsed patients have a good prognosis, with remarkably longer survival than refractory subjects.[90] There is no consensus on criteria to start rescue therapy in relapsing subjects.[91] Cardiac progression should not be awaited, because it is associated with shorter survival.[90]
Relapsing patients can be treated by repeating upfront therapy, if
possible, although this is associated with shorter time to retreatment
without reduction in overall survival.[92] When this
is not possible, relapsed patients should be treated as subjects
failing to respond to upfront therapy. A recent report defined that a
potential role of deferred ASCT in both a consolidation or relapse
setting in selected patients with cardiac AL who have achieved organ
responses.[93] Immunemodulatory drugs (IMiDs) are the
backbone of second-line therapy. Lenalidomide is able to overcome
resistance to alkylating agents, proteasome inhibitors, and
thalidomide.[94-99] However, this drug can cause worsening renal failure in patients with renal AL amyloidosis with significant proteinuria.[100] Lenalidomide combinations have been used also upfront with encouraging results.[97,98,101-104]
Pomalidomide is one of the most powerful agents in refractory AL
amyloidosis, being able to rescue patients refractory to alkylators,
first- and second-generation proteasome inhibitors, and lenalidomide.[105-107]
Hematologic response to pomalidomide is obtain rapidly, in a median
time of 1 months, and is observed in more than 60% of patients.[107]
Complete responses are relatively rare with IMiDs in pre-treated
patients. However, the use of IMiDs could result in long
progression-free intervals and survival rates among patients with AL
amyloidosis.[108] Newer agents have been tested in
the relapsed / refractory setting. The proteasome inhibitor carfilzomib
yielded a hematologic response rate of 63% (CR 12%).[109]
In this study, 39% of patients had NT-proBNP progression, which was
clinically relevant in 18% of cases. The cardiac toxicity of
carfilzomib is a cause of concern in AL amyloidosis. The oral
proteasome inhibitor ixazomib proved active in per-treated patients,
particularly in those who were not previously exposed to bortezomib,
and is currently being tested in a randomized phase III trial in
relapsed / refractory patients (NCT01659658).[110]
Thus, ixazomib seems particularly suitable for upfront combinations,
allowing oral proteasome inhibitor-based regimens. Indeed, 2 trials of
ixazomib, cyclophosphamide and dexamethasone (NCT03236792, NCT01864018)
are ongoing in the upfront setting. Daratumumab is one of the most
promising new agents for the treatment of patients with AL amyloidosis.
A recently published series of previously treated subjects who received
daratumumab reported a rapid (median 1 months) hematologic response in
76% of patients with 36% CRs.[111] In the 2017
American Society of hematology meeting, two different abstracts
reported the preliminary data of prospective ongoing clinical trials
about the use of daratumumab in relapse/refractory setting.[112,113]
Daratumumab will likely be moved to upfront therapy in combination with
proteasome inhibitor-based regimens in the near future. Indeed, a phase
III randomized trial of daratumumab in combination with CyBorD vs.
CyBorD alone in newly-diagnosed patients will be opened shortly
(NCT03201965).
Interfering with amyloidogenesis and organ damage and targeting the amyloid deposits.
New, non-hematologic, approaches specifically targeting steps that are
downstream in the pathogenic cascade are emerging as supplements of
anti-plasma cell therapy, given in combination with chemotherapy or
after achievement of hematologic response. Following the observation of
the efficacy of the anthracycline 4’-iodo-4’-deoxy-doxorubicin on
amyloidogenesis in vitro and reports of clinical improvements in
subjects with AL amyloidosis, related non-cytotoxic and non-cardiotoxic
compounds were investigated.[114-118] Amongst them, doxycycline was able to disrupt amyloid fibrils in transgenic mouse models of systemic amyloidosis,[119,120] and protected the C. elegans model from the effects of cardiotoxic amyloid light chains.[26]
In a retrospective case-control study the administration of doxycycline
as antibiotic prophylaxis during chemotherapy for AL amyloidosis
reduced early mortality, resulting in higher response rates and
survival improvement.[121] A phase III trial of
chemotherapy with or without doxycycline is being designed. Polyphenols
can redirect amyloidogenic polypeptides into unstructured, off-pathway
oligomers.[122] Amongst them
epigallocatechin-3-gallate was tested (EGCG) showed promising
activity on cardiac AL amyloidosis in case reports and retrospective
series.[123-125] In a phase II trial, EGCG was well tolerated and in some patients a decrease in albuminuria was observed.[126]
The
amyloid deposits are natural targets of novel therapies. United Kingdom
investigators designed a compound CPHPC that avidly binds to serum
amyloid P component (SAP) a protein that coats the amyloid fibrils
protecting them from degradation. This compound is used to remove SAP
from the bloodstream[127] before the administration
of an anti-SAP antibody that promotes a complement-dependent,
macrophage-derived reaction that removes visceral amyloid deposits.[128] This combination approach was tested in a pilot clinical study,[129]
and a trial in patients with cardiac AL amyloidosis is ongoing
(NCT03044353). The report of the ability of this approach to induce
organ response measured with validated criteria is eagerly awaited.
Antibodies directly targeting the amyloid deposits have also been
developed. One of them, 11-1F4, has been tested in phase I clinical
trial, showing promising organ response in an interim analysis.[130] Those data were recently updated at the last ASH meeting.[131]
A different antibody, NEOD001 is currently in the most advanced stage
of clinical development. In a phase I/II study in patients with AL
amyloidosis who had completed chemotherapy, cardiac and renal response
rates were 57% and 60%, respectively.[132] Organ response to NEOD001 was independent of rate and depth of previous hematologic response.[133]
Two phase III randomized, placebo-controlled trials of NEOD001 combined
with bortezomib-based chemotherapy in newly-diagnosed patients
(NCT02312206), and as single agent in subjects who completed
chemotherapy (NCT02632786) have recently completed enrollment and
results are eagerly awaited.
Supportive therapy.
Supportive treatment is vital in patients with AL amyloidosis, in order
to sustain organ function while chemotherapy takes effect, and to
improve quality of life. However, treatment of concomitant heart
failure and nephrotic syndrome in patients who often have concomitant
involvement of the autonomic nervous system is extremely complicated,
and should be done under close supervisions of specialized
cardiologists and nephrologists who have experience in the treatment of
patients with systemic amyloidosis. In some patients, asymptomatic
involvement of the autonomic nervous system[134]
could lead to overt, often severe hypotension when treatment with
angiotensin-converting enzyme inhibitors is established. This therapy
should be considered with caution and at the lowest effective dose. The
development of a significant peripheral edema requires diuretics
associated with dietary sodium restriction. Patients weigh themselves
daily, and diuretic dosing should be titrated accordingly. It should be
kept in mind that in patients with heat involvement cardiac function is
preload dependent and reduction of intravascular volume should be
avoided. Patients with recurrent arrhythmic syncope may benefit from
pacemaker implantation; whereas, the use of implantable ICD is
controversial. Gabapentin or pregabalin can be used to control
neuropathic pain and octreotide can control diarrhea. Nutritional
status is also frequently compromised in AL amyloidosis, independently
affecting quality of life assessment.[135-137] Nutritional counseling is effective in improving mental quality of life and is associated with better survival.[138]
Cardiac and renal transplant can be considered in patients who attain
CR but are dialysis dependent or have persistent, severe heart failure.
Moreover, young patients with isolated, advanced cardiac involvement
may be considered for heart transplant followed by effective
chemotherapy aiming at rapidly achieving CR.
Conclusions
Despite
the recent advances the management of AL amyloidosis remains highly
challenging and characterized by still unmet needs. The appropriate
management of AL amyloidosis requires 1) early diagnosis, 2) correct
typing, 3) accurate risk stratification and effective therapy guided by
frequent careful assessment of response. Early diagnosis lies in the
hands of general hematologists who are responsible for the follow-up of
patients with MGUS. The onset of cardiac and renal involvement by AL
amyloidosis in these subjects can be detected at a pre-symptomatic
stage with simple markers, albuminuria and NT-proBNP, that should be
part of the follow-up panel of patients with MGUS and abnormal FLC
ratio. Amyloid typing is mandatory but requires advanced technology
that needs to be concentrated at referral centers. The lack of
controlled prospective studies and the importance of a critical level
of expertise in specific and supportive therapy, also requires referral
of patients to specialized centers whenever possible. Coordinated
national networks are vital in sharing knowledge at rendering it
accessible to patients. In the near future, the availability of newer
powerful anti-plasma cell drugs, combined with anti-amyloid agent will
hopefully further improve the outcome of patients with AL amyloidosis.
Still clinical practice and research cannot be disconnected in this
complex and dreadful disease.
.
References
- Palladini G, Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood. 2016;128(2):159-168. https://doi.org/10.1182/blood-2016-01-629790 PMid:27053535

- Merlini G, Stone M. Dangerous small B-cell clones. Blood. 2006;108(8):2520-2530. https://doi.org/10.1182/blood-2006-03-001164 PMid:16794250
- Merlini
G. AL amyloidosis: from molecular mechanisms to targeted therapies.
Hematology Am Soc Hematol Educ Program. 2017;2017(1):1-12.
- Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-2654. https://doi.org/10.1016/S0140-6736(15)01274-X
- Muchtar
E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL
amyloidosis between 2000 and 2014: cracking the glass ceiling of early
death. Blood. 2017;129(15):2111-2119. https://doi.org/10.1182/blood-2016-11-751628 PMid:28126928
- Bochtler
T, Hegenbart U, Cremer F, et al. Evaluation of the cytogenetic
aberration pattern in amyloid light chain amyloidosis as compared with
monoclonal gammopathy of undetermined significance reveals common
pathways of karyotypic instability. Blood. 2008;111(9):4700-4705. https://doi.org/10.1182/blood-2007-11-122101 PMid:18305218
- Bochtler
T, Hegenbart U, Kunz C, et al. Translocation t(11;14) is associated
with adverse outcome in patients with newly diagnosed AL amyloidosis
when treated with bortezomib-based regimens. J Clin Oncol.
2015;33(12):1371-1378. https://doi.org/10.1200/JCO.2014.57.4947 PMid:25779559
- Muchtar
E, Dispenzieri A, Kumar SK, et al. Interphase fluorescence in situ
hybridization in untreated AL amyloidosis has an independent prognostic
impact by abnormality type and treatment category. Leukemia.
2017;31(7):1562-1569. https://doi.org/10.1038/leu.2016.369 PMid:27904139
- Muchtar
E, Dispenzieri A, Kumar SK, et al. Immunoparesis status in
immunoglobulin light chain amyloidosis at diagnosis affects response
and survival by regimen type. Haematologica. 2016;101(9):1102-1109. https://doi.org/10.3324/haematol.2016.147041 PMid:27479823 PMCid:PMC5060027
- Bochtler
T, Hegenbart U, Kunz C, et al. Prognostic impact of cytogenetic
aberrations in AL amyloidosis patients after high-dose melphalan: a
long-term follow-up study. Blood. 2016;128(4):594-602. https://doi.org/10.1182/blood-2015-10-676361 PMid:27257181
- Bochtler
T, Hegenbart U, Kunz C, et al. Gain of chromosome 1q21 is an
independent adverse prognostic factor in light chain amyloidosis
patients treated with melphalan/dexamethasone. Amyloid.
2014;21(1):9-17. https://doi.org/10.3109/13506129.2013.854766 PMid:24455967
- Paiva
B, Martinez-Lopez J, Corchete LA, et al. Phenotypic, transcriptomic,
and genomic features of clonal plasma cells in light-chain amyloidosis.
Blood. 2016;127(24):3035-3039. https://doi.org/10.1182/blood-2015-10-673095 PMid:27069257
- da
Silva Filho MI, Försti A, Weinhold N, et al. Genome-wide association
study of immunoglobulin light chain amyloidosis in three patient
cohorts: comparison with myeloma. Leukemia. 2017;31(8):1735-1742. https://doi.org/10.1038/leu.2016.387 PMid:28025584
- Sitia
R, Palladini G, Merlini G. Bortezomib in the treatment of AL
amyloidosis: targeted therapy? Haematologica. 2007;92(10):1302-1307. https://doi.org/10.3324/haematol.12136 PMid:18024367
- Bianchi
G, Oliva L, Cascio P, et al. The proteasome load versus capacity
balance determines apoptotic sensitivity of multiple myeloma cells to
proteasome inhibition. Blood. 2009;113(13):3040-3049. https://doi.org/10.1182/blood-2008-08-172734 PMid:19164601
- Oliva
L, Orfanelli U, Resnati M, et al. The amyloidogenic light chain is a
stressor that sensitizes plasma cells to proteasome inhibitor toxicity.
Blood. 2017;129(15):2132-2142. https://doi.org/10.1182/blood-2016-08-730978 PMid:28130214
- Comenzo
R, Zhang Y, Martinez C, Osman K, Herrera G. The tropism of organ
involvement in primary systemic amyloidosis: contributions of Ig V(L)
germ line gene use and clonal plasma cell burden. Blood.
2001;98(3):714-720. https://doi.org/10.1182/blood.V98.3.714 PMid:11468171
- Perfetti
V, Casarini S, Palladini G, et al. Analysis of V(lambda)-J(lambda)
expression in plasma cells from primary (AL) amyloidosis and normal
bone marrow identifies 3r (lambdaIII) as a new amyloid-associated
germline gene segment. Blood. 2002;100(3):948-953. https://doi.org/10.1182/blood-2002-01-0114 PMid:12130507
- Abraham
R, Geyer S, Price-Troska T, et al. Immunoglobulin light chain variable
(V) region genes influence clinical presentation and outcome in light
chain-associated amyloidosis (AL). Blood. 2003;101(10):3801-3808. https://doi.org/10.1182/blood-2002-09-2707 PMid:12515719
- Kourelis
TV, Dasari S, Theis JD, et al. Clarifying immunoglobulin gene usage in
systemic and localized immunoglobulin light-chain amyloidosis by mass
spectrometry. Blood. 2017;129(3):299-306. https://doi.org/10.1182/blood-2016-10-743997 PMid:27856462
- Perfetti
V, Palladini G, Casarini S, et al. The repertoire of ? light chains
causing predominant amyloid heart involvement and identification of a
preferentially involved germline gene, IGLV1-44. Blood.
2012;119(1):144-150. https://doi.org/10.1182/blood-2011-05-355784 PMid:22067386
- Palladini
G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic
peptide is a sensitive marker of myocardial dysfunction in AL
amyloidosis. Circulation. 2003;107(19):2440-2445. https://doi.org/10.1161/01.CIR.0000068314.02595.B2 PMid:12719281
- Palladini
G, Lavatelli F, Russo P, et al. Circulating amyloidogenic free light
chains and serum N-terminal natriuretic peptide type B decrease
simultaneously in association with improvement of survival in AL.
Blood. 2006;107(10):3854-3858. https://doi.org/10.1182/blood-2005-11-4385 PMid:16434487
- Palladini
G, Barassi A, Klersy C, et al. The combination of high-sensitivity
cardiac troponin T (hs-cTnT) at presentation and changes in N-terminal
natriuretic peptide type B (NT-proBNP) after chemotherapy best predicts
survival in AL amyloidosis. Blood. 2010;116(18):3426-3430. https://doi.org/10.1182/blood-2010-05-286567 PMid:20644111
- Liao
R, Jain M, Teller P, et al. Infusion of light chains from patients with
cardiac amyloidosis causes diastolic dysfunction in isolated mouse
hearts. Circulation. 2001;104(14):1594-1597 PMid:11581134
- Diomede
L, Rognoni P, Lavatelli F, et al. A Caenorhabditis elegans-based assay
recognizes immunoglobulin light chains causing heart amyloidosis.
Blood. 2014;123(23):3543-3552.
https://doi.org/10.1182/blood-2013-10-525634 PMid:24665135 PMCid:PMC4047494 - Mishra
S, Guan J, Plovie E, et al. Human amyloidogenic light chain proteins
result in cardiac dysfunction, cell death, and early mortality in
zebrafish. Am J Physiol Heart Circ Physiol. 2013;305(1):H95-103. https://doi.org/10.1152/ajpheart.00186.2013 PMid:23624626 PMCid:PMC3727100
- Lavatelli
F, Imperlini E, Orru S, et al. Novel mitochondrial protein interactors
of immunoglobulin light chains causing heart amyloidosis. FASEB J.
2015;29(11):4614-4628. https://doi.org/10.1096/fj.15-272179 PMid:26220173
- Oberti
L, Rognoni P, Barbiroli A, et al. Concurrent structural and biophysical
traits link with immunoglobulin light chains amyloid propensity. Sci
Rep. 2017;7(1):16809. https://doi.org/10.1038/s41598-017-16953-7 PMid:29196671 PMCid:PMC5711917
- Perlini
S, Salinaro F, Musca F, et al. Prognostic value of depressed midwall
systolic function in cardiac light-chain amyloidosis. J Hypertens.
2014;32(5):1121-1131; discussion 1131. https://doi.org/10.1097/HJH.0000000000000120 PMid:24509117
- Buss
SJ, Emami M, Mereles D, et al. Longitudinal left ventricular function
for prediction of survival in systemic light-chain amyloidosis:
incremental value compared with clinical and biochemical markers. J Am
Coll Cardiol. 2012;60(12):1067-1076. https://doi.org/10.1016/j.jacc.2012.04.043 PMid:22883634
- Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-144. https://doi.org/10.1007/s10741-014-9470-7 PMid:25549885
- Park
MA, Padera RF, Belanger A, et al. 18F-Florbetapir Binds Specifically to
Myocardial Light Chain and Transthyretin Amyloid Deposits:
Autoradiography Study. Circ Cardiovasc Imaging. 2015;8(8). https://doi.org/10.1161/CIRCIMAGING.114.002954
- Gillmore
JD, Maurer MS, Falk RH, et al. Nonbiopsy Diagnosis of Cardiac
Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-2412. https://doi.org/10.1161/CIRCULATIONAHA.116.021612 PMid:27143678
- Merlini
G, Palladini G. Differential diagnosis of monoclonal gammopathy of
undetermined significance. Hematology Am Soc Hematol Educ Program.
2012;2012:595-603.
- Lousada
I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light Chain Amyloidosis:
Patient Experience Survey from the Amyloidosis Research Consortium. Adv
Ther. 2015;32(10):920-928. https://doi.org/10.1007/s12325-015-0250-0 PMid:26498944 PMCid:PMC4635176
- Kourelis
TV, Kumar SK, Go RS, et al. Immunoglobulin light chain amyloidosis is
diagnosed late in patients with preexisting plasma cell dyscrasias. Am
J Hematol. 2014;89(11):1051-1054.https://doi.org/10.1002/ajh.23827 PMid:25111004
- Merlini
G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an
update for treating physicians. Blood. 2013;121(26):5124-5130. https://doi.org/10.1182/blood-2013-01-453001 PMid:23670179
- Foli
A, Palladini G, Caporali R, et al. The role of minor salivary gland
biopsy in the diagnosis of systemic amyloidosis: results of a
prospective study in 62 patients. Amyloid. 2011;18 Suppl 1:80-82. https://doi.org/10.3109/13506129.2011.574354029 PMid:21838441
- Fernandez
de Larrea C, Verga L, Morbini P, et al. A practical approach to the
diagnosis of systemic amyloidoses. Blood. 2015;125(14):2239-2244. https://doi.org/10.1182/blood-2014-11-609883 PMid:25636337
- Muchtar
E, Dispenzieri A, Lacy MQ, et al. Overuse of organ biopsies in
immunoglobulin light chain amyloidosis (AL): the consequence of failure
of early recognition. Ann Med. 2017;49(7):545-551. https://doi.org/10.1080/07853890.2017.1304649 PMid:28271734
- Anesi
E, Palladini G, Perfetti V, Arbustini E, Obici L, Merlini G.
Therapeutic advances demand accurate typing of amyloid deposits. Am J
Med. 2001;111(3):243-244. https://doi.org/10.1016/S0002-9343(01)00774-4
- Lachmann
H, Booth D, Booth S, et al. Misdiagnosis of hereditary amyloidosis as
AL (primary) amyloidosis. N Engl J Med. 2002;346(23):1786-1791. https://doi.org/10.1056/NEJMoa013354 PMid:12050338
- Comenzo
R, Zhou P, Fleisher M, Clark B, Teruya-Feldstein J. Seeking confidence
in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients
can have both monoclonal gammopathies and hereditary amyloid proteins.
Blood. 2006;107(9):3489-3491. https://doi.org/10.1182/blood-2005-10-4148 PMid:16439680
- Satoskar
A, Burdge K, Cowden D, Nadasdy G, Hebert L, Nadasdy T. Typing of
amyloidosis in renal biopsies: diagnostic pitfalls. Arch Pathol Lab
Med. 2007;131(6):917-922. PMid:17550319
- Satoskar
AA, Efebera Y, Hasan A, et al. Strong transthyretin immunostaining:
potential pitfall in cardiac amyloid typing. Am J Surg Pathol.
2011;35(11):1685-1690. https://doi.org/10.1097/PAS.0b013e3182263d74 PMid:21945954 PMCid:PMC4061151
- Schönland
SO, Hegenbart U, Bochtler T, et al. Immunohistochemistry in the
classification of systemic forms of amyloidosis: a systematic
investigation of 117 patients. Blood. 2012;119(2):488-493. https://doi.org/10.1182/blood-2011-06-358507 PMid:22106346
- Vrana
J, Gamez J, Madden B, Theis J, Bergen Hr, Dogan A. Classification of
amyloidosis by laser microdissection and mass spectrometry-based
proteomic analysis in clinical biopsy specimens. Blood.
2009;114(24):4957-4959. https://doi.org/10.1182/blood-2009-07-230722 PMid:19797517
- Brambilla
F, Lavatelli F, Di Silvestre D, et al. Reliable typing of systemic
amyloidoses through proteomic analysis of subcutaneous adipose tissue.
Blood. 2012;119(8):1844-1847. https://doi.org/10.1182/blood-2011-07-365510 PMid:21917755
- Katzmann
J, Kyle R, Benson J, et al. Screening panels for detection of
monoclonal gammopathies. Clin Chem. 2009;55(8):1517-1522. https://doi.org/10.1373/clinchem.2009.126664 PMid:19520758 PMCid:PMC3773468
- Bochtler
T, Hegenbart U, Heiss C, et al. Evaluation of the serum-free light
chain test in untreated patients with AL amyloidosis. Haematologica.
2008;93(3):459-462. https://doi.org/10.3324/haematol.11687 PMid:18287137
- Palladini
G, Russo P, Bosoni T, et al. Identification of amyloidogenic light
chains requires the combination of serum-free light chain assay with
immunofixation of serum and urine. Clin Chem. 2009;55(3):499-504. https://doi.org/10.1373/clinchem.2008.117143 PMid:19131635
- Palladini
G, Jaccard A, Milani P, et al. Circulating free light chain measurement
in the diagnosis, prognostic assessment and evaluation of response of
AL amyloidosis: comparison of Freelite and N latex FLC assays. Clin
Chem Lab Med. 2017. https://doi.org/10.1515/cclm-2016-1024 PMid:28343171
- Dispenzieri
A, Gertz M, Kyle R, et al. Serum cardiac troponins and N-terminal
pro-brain natriuretic peptide: a staging system for primary systemic
amyloidosis. J Clin Oncol. 2004;22(18):3751-3757. https://doi.org/10.1200/JCO.2004.03.029 PMid:15365071
- Wechalekar
AD, Schonland SO, Kastritis E, et al. A European collaborative study of
treatment outcomes in 346 patients with cardiac stage III AL
amyloidosis. Blood. 2013;121(17):3420-3427. https://doi.org/10.1182/blood-2012-12-473066 PMid:23479568
- Kourelis
TV, Kumar SK, Gertz MA, et al. Coexistent multiple myeloma or increased
bone marrow plasma cells define equally high-risk populations in
patients with immunoglobulin light chain amyloidosis. J Clin Oncol.
2013;31(34):4319-4324. https://doi.org/10.1200/JCO.2013.50.8499 PMid:24145344 PMCid:PMC4881366
- Dittrich
T, Bochtler T, Kimmich C, et al. AL amyloidosis patients with low
amyloidogenic free light chain levels at first diagnosis have an
excellent prognosis. Blood. 2017;130(5):632-642. https://doi.org/10.1182/blood-2017-02-767475 PMid:28550043
- Milani
P, Basset M, Russo F, Foli A, Merlini G, Palladini G. Patients with
light-chain amyloidosis and low free light-chain burden have distinct
clinical features and outcome. Blood. 2017;130(5):625-631. https://doi.org/10.1182/blood-2017-02-767467 PMid:28546143
- Kumar
S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for
light chain amyloidosis incorporating cardiac biomarkers and serum free
light chain measurements. J Clin Oncol. 2012;30(9):989-995. https://doi.org/10.1200/JCO.2011.38.5724 PMid:22331953 PMCid:PMC3675680
- Palladini
G, Hegenbart U, Milani P, et al. A staging system for renal outcome and
early markers of renal response to chemotherapy in AL amyloidosis.
Blood. 2014;124(15):2325-2332. https://doi.org/10.1182/blood-2014-04-570010 PMid:25115890
- D'Souza
A, Dispenzieri A, Wirk B, et al. Improved Outcomes After Autologous
Hematopoietic Cell Transplantation for Light Chain Amyloidosis: A
Center for International Blood and Marrow Transplant Research Study. J
Clin Oncol. 2015;33(32):3741-3749. https://doi.org/10.1200/JCO.2015.62.4015 PMid:26371138 PMCid:PMC4737858
- Rajkumar
SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working
Group updated criteria for the diagnosis of multiple myeloma. Lancet
Oncol. 2014;15(12):e538-e548. https://doi.org/10.1016/S1470-2045(14)70442-5
- Dispenzieri
A, Seenithamby K, Lacy MQ, et al. Patients with immunoglobulin light
chain amyloidosis undergoing autologous stem cell transplantation have
superior outcomes compared with patients with multiple myeloma: a
retrospective review from a tertiary referral center. Bone Marrow
Transplant. 2013;48(10):1302-1307. https://doi.org/10.1038/bmt.2013.53 PMid:23604010
- Palladini
G, Dispenzieri A, Gertz MA, et al. New criteria for response to
treatment in immunoglobulin light chain amyloidosis based on free light
chain measurement and cardiac biomarkers: impact on survival outcomes.
J Clin Oncol. 2012;30(36):4541-4549. https://doi.org/10.1200/JCO.2011.37.7614 PMid:23091105
- Merlini
G, Lousada I, Ando Y, et al. Rationale, application and clinical
qualification for NT-proBNP as a surrogate end point in pivotal
clinical trials in patients with AL amyloidosis. Leukemia.
2016;30(10):1979-1986. https://doi.org/10.1038/leu.2016.191 PMid:27416985 PMCid:PMC5056962
- Sidana
S, Tandon, N., Dispenzieri, A., Gertz, M. A., Buadi, F. K., Lacy, M.
Q., Dingli, D., Fonder, A., Hayman, S. R., Hobbs, M., Gonsalves, W. I.,
Hwa, Y. L., Kapoor, P., Kyle, R. A., Leung, N., Go, R. S., Lust, J. A.,
Russell, S. J., Zeldenrust, S. R., Binder, M., Rajkumar, S. V., &
Kumar, S. K. Composite Organ and Hematologic Response Model Risk
Stratifies Patients with Light Chain Amyloidosis. Blood. 2017;130
(Suppl 1), 3046 .
- Muchtar
E, Dispenzieri, A., Lacy, M. Q., Leung, N., Buadi, F. K., Grogan, M.,
Hayman, S. R., Kapoor, P., Hwa, Y. L., Fonder, A., Hobbs, M.,
Chakraborty, R., Gonsalves, W. I., Kourelis, T., Warsame, R., Russell,
S. J., Lust, J. A., Lin, Y., Go, R. S., Zeldenrust, S., Kyle, R. A.,
Rajkumar, S. V., Kumar, S. K., & Gertz, M. A. . Depth of Organ
Response in Newly Diagnosed AL Amyloidosis Is Associated with Improved
Survival: Improved Outcome Discrimination with Graded Organ
Response. Blood. 2017;130 (Suppl 1), 3154 .
- Sanchorawala
V, Sun F, Quillen K, Sloan JM, Berk JL, Seldin DC. Long-term outcome of
patients with AL amyloidosis treated with high-dose melphalan and stem
cell transplantation: 20-year experience. Blood.
2015;126(20):2345-2347. https://doi.org/10.1182/blood-2015-08-662726 PMid:26443620
- Gertz
MA, Lacy MQ, Dispenzieri A, et al. Refinement in patient selection to
reduce treatment-related mortality from autologous stem cell
transplantation in amyloidosis. Bone Marrow Transplant.
2013;48(4):557-561. https://doi.org/10.1038/bmt.2012.170 PMid:22964596
- Dispenzieri
A, Buadi F, Kumar SK, et al. Treatment of Immunoglobulin Light Chain
Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy
(mSMART) Consensus Statement. Mayo Clin Proc. 2015;90(8):1054-1081. https://doi.org/10.1016/j.mayocp.2015.06.009 PMid:26250727
- Cibeira
MT, Sanchorawala V, Seldin DC, et al. Outcome of AL amyloidosis after
high-dose melphalan and autologous stem cell transplantation: long-term
results in a series of 421 patients. Blood. 2011;118(16):4346-4352. https://doi.org/10.1182/blood-2011-01-330738 PMid:21828140 PMCid:PMC3204906
- Landau
H, Smith M, Landry C, et al. Long-term event-free and overall survival
after risk-adapted melphalan and SCT for systemic light chain
amyloidosis. Leukemia. 2017;31(1):136-142. https://doi.org/10.1038/leu.2016.229 PMid:27560108 PMCid:PMC5220129
- Hwa
YL, Kumar SK, Gertz MA, et al. Induction therapy pre-autologous stem
cell transplantation in immunoglobulin light chain amyloidosis: a
retrospective evaluation. Am J Hematol. 2016;91(10):984-988. https://doi.org/10.1002/ajh.24453 PMid:27341539
- Palladini
G, Perfetti V, Obici L, et al. Association of melphalan and high-dose
dexamethasone is effective and well tolerated in patients with AL
(primary) amyloidosis who are ineligible for stem cell transplantation.
Blood. 2004;103(8):2936-2938. https://doi.org/10.1182/blood-2003-08-2788 PMid:15070667
- Palladini
G, Russo P, Nuvolone M, et al. Treatment with oral melphalan plus
dexamethasone produces long-term remissions in AL amyloidosis. Blood.
2007;110(2):787-788. https://doi.org/10.1182/blood-2007-02-076034 PMid:17606766
- Palladini
G, Milani P, Foli A, et al. Oral melphalan and dexamethasone grants
extended survival with minimal toxicity in AL amyloidosis: long-term
results of a risk-adapted approach. Haematologica. 2014;99(4):743-750. https://doi.org/10.3324/haematol.2013.095463 PMid:24213149 PMCid:PMC3971085
- Jaccard
A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan
plus dexamethasone for AL amyloidosis. N Engl J Med.
2007;357(11):1083-1093. https://doi.org/10.1056/NEJMoa070484 PMid:17855669
- Kastritis
E, Wechalekar A, Dimopoulos M, et al. Bortezomib with or without
dexamethasone in primary systemic (light chain) amyloidosis. J Clin
Oncol. 2010;28(6):1031-1037. https://doi.org/10.1200/JCO.2009.23.8220 PMid:20085941
- Reece
D, Sanchorawala V, Hegenbart U, et al. Weekly and twice-weekly
bortezomib in patients with systemic AL amyloidosis: results of a phase
1 dose-escalation study. Blood. 2009;114(8):1489-1497. https://doi.org/10.1182/blood-2009-02-203398 PMid:19498019
- Reece
DE, Hegenbart U, Sanchorawala V, et al. Efficacy and safety of
once-weekly and twice-weekly bortezomib in patients with relapsed
systemic AL amyloidosis: results of a phase 1/2 study. Blood.
2011;118(4):865-873. https://doi.org/10.1182/blood-2011-02-334227 PMid:21562045
- Reece
DE, Hegenbart U, Sanchorawala V, et al. Long-term follow-up from a
phase 1/2 study of single-agent bortezomib in relapsed systemic AL
amyloidosis. Blood. 2014;124(16):2498-2506. https://doi.org/10.1182/blood-2014-04-568329 PMid:25202139 PMCid:PMC4199951
- Palladini
G, Sachchithanantham S, Milani P, et al. A European collaborative study
of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment
of systemic AL amyloidosis. Blood. 2015;126(5):612-615. https://doi.org/10.1182/blood-2015-01-620302 PMid:25987656
- Palladini
G, Milani P, Foli A, et al. Melphalan and dexamethasone with or without
bortezomib in newly diagnosed AL amyloidosis: a matched case-control
study on 174 patients. Leukemia. 2014;28(12):2311-2316. https://doi.org/10.1038/leu.2014.227 PMid:25059496
- Venner
CP, Gillmore JD, Sachchithanantham S, et al. A matched comparison of
cyclophosphamide, bortezomib and dexamethasone (CVD) versus
risk-adapted cyclophosphamide, thalidomide and dexamethasone (CTD) in
AL amyloidosis. Leukemia. 2014;28(12):2304-2310. https://doi.org/10.1038/leu.2014.218 PMid:25027514
- Kastritis
E, Leleu X, Arnulf B, et al. A Randomized Phase III Trial of Melphalan
and Dexamethasone (MDex) Versus Bortezomib, Melphalan and Dexamethasone
(BMDex) for Untreated Patients with AL Amyloidosis. Blood. 2016;128(22).
- Kastritis
E, Gavriatopoulou M, Roussou M, et al. Addition of cyclophosphamide and
higher doses of dexamethasone do not improve outcomes of patients with
AL amyloidosis treated with bortezomib. Blood Cancer J. 2017;7(6):e570.
https://doi.org/10.1038/bcj.2017.47 PMid:28622303 PMCid:PMC5520394
- Sidana
S, Tandon N, Gertz MA, et al. Impact of prior melphalan exposure on
stem cell collection in light chain amyloidosis. Bone Marrow
Transplant. 2017. https://doi.org/10.1038/s41409-017-0020-5
- Palladini
G, Milani P, Merlini G. Novel strategies for the diagnosis and
treatment of cardiac amyloidosis. Expert review of cardiovascular
therapy. 2015;13(11):1195-1211. https://doi.org/10.1586/14779072.2015.1093936 PMid:26496239
- Manwani
R, Foard D, Mahmood S, et al. Rapid hematological responses improve
outcomes in patients with very advanced (Stage IIIb) cardiac
immunoglobulin light chain amyloidosis. Haematologica. 2018. https://doi.org/10.3324/haematol.2017.178095 PMid:29305414
- Palladini
G, Milani P, Foli A, et al. Presentation and outcome with second line
treatment in AL amyloidosis previously sensitive to non-transplant
therapies. Blood. 2017.
- Milani
P, Gertz MA, Merlini G, Dispenzieri A. Attitudes about when and how to
treat patients with AL amyloidosis: an international survey. Amyloid.
2017:1-4. https://doi.org/10.1080/13506129.2017.1370421
- Tandon
N, Sidana S, Gertz MA, et al. Treatment Patterns and Outcome Following
Initial Relapse or Refractory Disease in Patients with Systemic Light
Chain Amyloidosis. Am J Hematol. 2017. https://doi.org/10.1002/ajh.24723
- Manwani
R, Hegenbart, U., Foard, D., Mahmood, S., Sachchithanantham, S., Yong,
K., Popat, R., Kyriakou, C., Rabin, N., Fontana, M., Whelan, C.,
Lachmann, H., Lane, T., Quarta, C., Gillmore, J., Dittrich, T.,
Kimmich, C., Hawkins, P., Schönland, S., & Wechalekar, A. D. Safety
and Efficacy of Deferred Autologous Stem Cell Transplantation in
Patients with Systemic AL Amyloidosis with Significant Cardiac
Involvement at Presentation. Blood. 2017;130 (Suppl 1), 1815.
- Dispenzieri
A, Lacy M, Zeldenrust S, et al. The activity of lenalidomide with or
without dexamethasone in patients with primary systemic amyloidosis.
Blood. 2007;109(2):465-470. https://doi.org/10.1182/blood-2006-07-032987 PMid:17008538
- Sanchorawala
V, Wright D, Rosenzweig M, et al. Lenalidomide and dexamethasone in the
treatment of AL amyloidosis: results of a phase 2 trial. Blood.
2007;109(2):492-496. https://doi.org/10.1182/blood-2006-07-030544 PMid:16960148
- Palladini
G, Russo P, Foli A, et al. Salvage therapy with lenalidomide and
dexamethasone in patients with advanced AL amyloidosis refractory to
melphalan, bortezomib, and thalidomide. Ann Hematol. 2011 .
- Kastritis
E, Terpos E, Roussou M, et al. A phase 1/2 study of lenalidomide with
low-dose oral cyclophosphamide and low-dose dexamethasone (RdC) in AL
amyloidosis. Blood. 2012;119(23):5384-5390. https://doi.org/10.1182/blood-2011-12-396903 PMid:22517904
- Kumar
SK, Hayman SR, Buadi FK, et al. Lenalidomide, cyclophosphamide, and
dexamethasone (CRd) for light-chain amyloidosis: long-term results from
a phase 2 trial. Blood. 2012;119(21):4860-4867. https://doi.org/10.1182/blood-2012-01-407791 PMid:22504925 PMCid:PMC3418771
- Mahmood
S, Venner CP, Sachchithanantham S, et al. Lenalidomide and
dexamethasone for systemic AL amyloidosis following prior treatment
with thalidomide or bortezomib regimens. Br J Haematol.
2014;166(6):842-848. https://doi.org/10.1111/bjh.12973 PMid:24930361
- Specter
R, Sanchorawala V, Seldin DC, et al. Kidney dysfunction during
lenalidomide treatment for AL amyloidosis. Nephrol Dial Transplant.
2011;26(3):881-886. https://doi.org/10.1093/ndt/gfq482 PMid:20693160 PMCid:PMC3108346
- Moreau
P, Jaccard A, Benboubker L, et al. Lenalidomide in combination with
melphalan and dexamethasone in patients with newly diagnosed AL
amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood.
2010;116(23):4777-4782. https://doi.org/10.1182/blood-2010-07-294405 PMid:20724537
- Sanchorawala
V, Patel JM, Sloan JM, Shelton AC, Zeldis JB, Seldin DC. Melphalan,
lenalidomide and dexamethasone for the treatment of immunoglobulin
light chain amyloidosis: results of a phase II trial. Haematologica.
2013;98(5):789-792. https://doi.org/10.3324/haematol.2012.075192 PMid:23144200 PMCid:PMC3640126
- Cibeira
MT, Oriol A, Lahuerta JJ, et al. A phase II trial of lenalidomide,
dexamethasone and cyclophosphamide for newly diagnosed patients with
systemic immunoglobulin light chain amyloidosis. Br J Haematol.
2015;170(6):804-813. https://doi.org/10.1111/bjh.13500 PMid:25974382
- Hegenbart
U, Bochtler T, Benner A, et al. Lenalidomide/ melphalan/dexamethasone
in newly diagnosed patients with immunoglobulin light chain
amyloidosis: results of a prospective phase 2 study with long-term
follow up. Haematologica. 2017;102(8):1424-1431. https://doi.org/10.3324/haematol.2016.163246 PMid:28522573 PMCid:PMC5541875
- Dispenzieri
A, Buadi F, Laumann K, et al. Activity of pomalidomide in patients with
immunoglobulin light-chain amyloidosis. Blood. 2012;119(23):5397-5404. https://doi.org/10.1182/blood-2012-02-413161 PMid:22493299 PMCid:PMC3369677
- Sanchorawala
V, Shelton AC, Lo S, Varga C, Sloan JM, Seldin DC. Pomalidomide and
dexamethasone in the treatment of AL amyloidosis: results of a phase 1
and 2 trial. Blood. 2016;128(8):1059-1062. https://doi.org/10.1182/blood-2016-04-710822 PMid:27381904
- Palladini
G, Milani P, Foli A, et al. A phase 2 trial of pomalidomide and
dexamethasone rescue treatment in patients with AL amyloidosis. Blood.
2017;129(15):2120-2123. https://doi.org/10.1182/blood-2016-12-756528 PMid:28130212
- Warsame
R, Laplant, B., Laumann, K., Kumar, S. K., Gertz, M. A., Kyle, R. A.,
Lacy, M. Q., Dingli, D., Leung, N., Buadi, F. K., Hayman, S. R.,
Kapoor, P., Hwa, Y. L., Fonder, A., Hobbs, M., Gonsalves, W. I.,
Kourelis, T., Russell, S. J., Zeldenrust, S., Lin, Y., Muchtar, E., Go,
R. S., Rajkumar, S. V., & Dispenzieri, A.. Long-Term Outcomes of
IMiD Based Trials in Patients with Immunoglobulin Light Chain
Amyloidosis (AL): A Pooled Analysis. Blood. 2017;130 (Suppl 1),
1833.
- Cohen
A, Landau H, Scott E, et al. Safety and Efficacy of Carfilzomib (CFZ)
in Previously-Treated Systemic Light-Chain (AL) Amyloidosis. Blood.
2016;128(22). PMCid:PMC5054698
- Sanchorawala
V, Palladini G, Kukreti V, et al. A phase 1/2 study of the oral
proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis.
Blood. 2017;130(5):597-605. https://doi.org/10.1182/blood-2017-03-771220 PMid:28550039
- Kaufman
GP, Schrier SL, Lafayette RA, Arai S, Witteles RM, Liedtke M.
Daratumumab yields rapid and deep hematologic responses in patients
with heavily pretreated AL amyloidosis. Blood. 2017. https://doi.org/10.1182/blood-2017-01-763599 PMCid:PMC5570682
- Sanchorawala
V, Sarosiek, S., Sloan, J. M., Brauneis, D., Migre, M. E., Mistark, M.,
Santos, S., Fennessey, S., & Shelton, A. C.. Safety and
Tolerability of Daratumumab in Patients with Relapsed Light Chain (AL)
Amyloidosis: Preliminary Results of a Phase II Study. Blood.
2017;130(Suppl 1), 507.
- Roussel
M, Stoppa, A., Perrot, A., Karlin, L., Arnulf, B., Macro, M., Huart,
A., Frenzel, L., Morel, P., Boyle, E., Dorvaux, V., Merlini, G.,
Palladini, G., Lavergne, D., Bridoux, F., & Jaccard, A. A
Prospective Phase II of Daratumumab in Previously-Treated Systemic
Light-Chain (AL) Amyloidosis. Blood. 2017;130(Suppl 1), 508.
- Merlini
G, Ascari E, Amboldi N, et al. Interaction of the anthracycline
4'-iodo-4'-deoxydoxorubicin with amyloid fibrils: inhibition of
amyloidogenesis. Proc Natl Acad Sci U S A. 1995;92(7):2959-2963. https://doi.org/10.1073/pnas.92.7.2959 PMid:7708755 PMCid:PMC42338
- Merlini
G, Anesi E, Garini P, et al. Treatment of AL amyloidosis with
4'-lodo-4'-deoxydoxorubicin: an update. Blood. 1999;93(3):1112-1113.
PMid:10025983
- Palha
JA, Ballinari D, Amboldi N, et al. 4'-Iodo-4'-deoxydoxorubicin disrupts
the fibrillar structure of transthyretin amyloid. Am J Pathol.
2000;156(6):1919-1925. https://doi.org/10.1016/S0002-9440(10)65065-1
- Cardoso
I, Merlini G, Saraiva MJ. 4'-iodo-4'-deoxydoxorubicin and tetracyclines
disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic
species: screening for TTR fibril disrupters. FASEB J.
2003;17(8):803-809. https://doi.org/10.1096/fj.02-0764com PMid:12724338
- Gertz
M, Lacy M, Dispenzieri A, et al. A multicenter phase II trial of
4'-iodo-4'deoxydoxorubicin (IDOX) in primary amyloidosis (AL). Amyloid.
2002;9(1):24-30. https://doi.org/10.3109/13506120209072441 PMid:12000194
- Cardoso
I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence
from studies in a FAP transgenic mice model. FASEB J.
2006;20(2):234-239. https://doi.org/10.1096/fj.05-4509com PMid:16449795
- Ward
JE, Ren R, Toraldo G, et al. Doxycycline reduces fibril formation in a
transgenic mouse model of AL amyloidosis. Blood.
2011;118(25):6610-6617. https://doi.org/10.1182/blood-2011-04-351643 PMid:21998211 PMCid:PMC3242721
- Wechalekar
AD, Whelan C. Encouraging impact of doxycycline on early mortality in
cardiac light chain (AL) amyloidosis. Blood Cancer J. 2017;7(3):e546.
Edwards CV, Gould J, Langer AL, et al. Interim analysis of the phase
1a/b study of chimeric fibril-reactive monoclonal antibody 11-1F4 in
patients with AL amyloidosis. Amyloid. 2017;24(supp1):58-59.
- Ehrnhoefer
DE, Bieschke J, Boeddrich A, et al. EGCG redirects amyloidogenic
polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol
Biol. 2008;15(6):558-566. https://doi.org/10.1038/nsmb.1437 PMid:18511942
- Hora
M, Carballo-Pacheco M, Weber B, et al. Epigallocatechin-3-gallate
preferentially induces aggregation of amyloidogenic immunoglobulin
light chains. Sci Rep. 2017;7:41515. https://doi.org/10.1038/srep41515 PMid:28128355 PMCid:PMC5269747
- Hunstein W. Epigallocathechin-3-gallate in AL amyloidosis: a new therapeutic option? Blood. 2007;110(6):2216. https://doi.org/10.1182/blood-2007-05-089243 PMid:17785589
- Mereles
D, Buss SJ, Hardt SE, Hunstein W, Katus HA. Effects of the main green
tea polyphenol epigallocatechin-3-gallate on cardiac involvement in
patients with AL amyloidosis. Clin Res Cardiol. 2010;99(8):483-490. https://doi.org/10.1007/s00392-010-0142-x PMid:20221615
- Meshitsuka
S, Shingaki S, Hotta M, et al. Phase 2 trial of daily, oral
epigallocatechin gallate in patients with light-chain amyloidosis. Int
J Hematol. 2017;105(3):295-308. https://doi.org/10.1007/s12185-016-2112-1 PMid:27815860
- Pepys
M, Herbert J, Hutchinson W, et al. Targeted pharmacological depletion
of serum amyloid P component for treatment of human amyloidosis.
Nature. 2002;417(6886):254-259. https://doi.org/10.1038/417254a PMid:12015594
- Gillmore
JD, Tennent GA, Hutchinson WL, et al. Sustained pharmacological
depletion of serum amyloid P component in patients with systemic
amyloidosis. Br J Haematol. 2010;148(5):760-767. https://doi.org/10.1111/j.1365-2141.2009.08036.x PMid:20064157
- Richards
DB, Cookson LM, Berges AC, et al. Therapeutic Clearance of Amyloid by
Antibodies to Serum Amyloid P Component. N Engl J Med.
2015;373(12):1106-1114. https://doi.org/10.1056/NEJMoa1504942 PMid:26176329
- Edwards
CV, Gould J, Langer AL, et al. Interim analysis of the phase 1a/b study
of chimeric fibril-reactive monoclonal antibody 11-1F4 in patients with
AL amyloidosis. Amyloid. 2017;24 (supp1):58-59.
- Edwards
CV, Gould, J., Langer, A.L., Mapara, M. Y., Radhakrishnan, J., Maurer,
M.S., Raza, S., Mears, J.G., Leng, S., Wall, J.S., Eisenberger, A.,
Solomon, A., & Lentzsch, S. Final Analysis of the Phase 1a/b Study
of Chimeric Fibril-Reactive Monoclonal Antibody 11-1F4 in Patients with
Relapsed or Refractory AL Amyloidosis. Blood. 2017;130 (Suppl 1), 509.
- Gertz
MA, Landau H, Comenzo RL, et al. First-in-Human Phase I/II Study of
NEOD001 in Patients With Light Chain Amyloidosis and Persistent Organ
Dysfunction. J Clin Oncol. 2016;34(10):1097-1103. https://doi.org/10.1200/JCO.2015.63.6530 PMid:26858336 PMCid:PMC5470113
- Gertz
M, Comenzo R, Landau H, et al. Patients with light chain amyloidosis
treated with neod001 achieve rapid organ responses that are independent
of previous plasma cell-directed therapies. Haematologica.
2017;102:3-3.
- Bernardi
L, Passino C, Porta C, Anesi E, Palladini G, Merlini G. Widespread
cardiovascular autonomic dysfunction in primary amyloidosis: does
spontaneous hyperventilation have a compensatory role against postural
hypotension? Heart. 2002;88(6):615-621. https://doi.org/10.1136/heart.88.6.615 PMid:12433892 PMCid:PMC1767452
- Caccialanza
R, Palladini G, Klersy C, et al. Nutritional status of outpatients with
systemic immunoglobulin light-chain amyloidosis. American Journal of
Clinical Nutrition. 2006;83(2):350-354. PMid:16469994
- Caccialanza
R, Palladini G, Klersy C, et al. Nutritional status independently
affects quality of life of patients with systemic immunoglobulin
light-chain (AL) amyloidosis. Ann Hematol. 2012;91(3):399-406. https://doi.org/10.1007/s00277-011-1309-x PMid:21826471
- Caccialanza
R, Palladini G, Klersy C, et al. Malnutrition at diagnosis predicts
mortality in patients with systemic immunoglobulin light-chain
amyloidosis independently of cardiac stage and response to treatment.
JPEN J Parenter Enteral Nutr. 2014;38(7):891-894. https://doi.org/10.1177/0148607113501328 PMid:24072737
- Caccialanza
R, Palladini G, Cereda E, et al. Nutritional counseling improves
quality of life and preserves body weight in systemic immunoglobulin
light-chain (AL) amyloidosis. Nutrition. 2015;31(10):1228-1234. https://doi.org/10.1016/j.nut.2015.04.011 PMid:26250487
- Kumar
S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for
light chain amyloidosis incorporating cardiac biomarkers and serum free
light chain measurements. J Clin Oncol. 2012;30(9):989-995. https://doi.org/10.1200/JCO.2011.38.5724 PMid:22331953 PMCid:PMC3675680
[TOP]