Naouel Guirat Dhouib,
Monia Ben Khaled, Monia Ouederni, Habib Besbes, Ridha Kouki, Fethi
Mellouli and Mohamed Bejaoui..
Pediatric Immuno-Hematology Unit, Bone Marrow Transplantation Center Tunis, Tunis, Tunisia.
Corresponding
author: Naouel Guirat Dhouib, Pediatric Immuno-Hematology Unit, Bone
Marrow Transplantation Center Tunis, Tunis, Tunisia. Tel: + 216 98
644165. E-mail:
nawel.guirat@yahoo.fr
Published: May 1, 2018
Received: March 6, 2018
Accepted: April 16, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018031 DOI
10.4084/MJHID.2018.031
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
β-thalassemia major (β-TM)
is among the most common hereditary disorders imposing high expenses on
health-care system worldwide. The patient's survival is dependent on
lifetime blood transfusion which leads to iron overload and its
toxicity in various organs including endocrine glands. This article
provides an overview of endocrine disorders in beta-TM patients. This
single center investigation enrolled 28 β-TM
patients (16 males, 12 females) regularly transfused with packed red
cell since early years of life. For each patient were determined: age,
sex, number of transfusions received, history of splenectomy and
anthropometric parameters. Evaluation of hormonal status including
growth, gonadal, thyroid, adrenal cortex, and parathyroid glands was
done for all patients. Dual-energy X-ray absorptiometry was used to
diagnose low bone mass. Assessment of iron overload status was
performed by measuring the serum ferritin concentration and the results
of magnetic resonance imaging T2*.
Growth retardation was found in 16 of the 28 studied patients
(57%).Thirteen among them had delayed puberty. Spontaneous puberty was
achieved in 16 cases. Growth hormone (GH) deficiency was found in 10
cases (35%). Seventeen among the studied patients (60%) developed
disorders of glucose homeostasis. Subclinical hypothyroidism was found
in six patients (21%). Intensive chelation therapy had allowed the
reversibility of this complication in five cases. Adrenal Insufficiency
was observed in 9 cases (32%). Hypoparathyroidism has occurred in one
case. Ten of the 28 studied patients had low bone mass (35%).
Twenty-three of the 28 studied patients (82%) had at least one
endocrine complication.
|
Introduction
Long-term transfusion regimen associated with optimal chelation therapy guided by magnetic resonance imaging (MRI T2*) technology has dramatically improved life expectancy in patients with β-TM.[1]
This hemoglobinopathy, which once incompatible with prolonged survival
has become a chronic disease compatible with prolonged survival.
Endocrine disorders are among the most common complications in multi-transfused β-thalassemia
major. They represent a leading cause of morbidity and have a
significant impact on the quality of life of patients suffering from
it.[2]To our knowledge; this is the first single center study reporting
endocrine disorders in Tunisian β-thalassemia major patients.
Patients and Methods
Twenty-eight
polytransfused thalassemia major patients older than ten years (19 ±
4,54) followed in the pediatric Immuno-hematology Department of the
Bone Marrow Transplantation during a 13-year period were enrolled in
the study. The diagnosis of β-TM was established by the finding of
microcytic hypochromic anemia, hemoglobin analysis before blood
transfusion that revealed increased amounts of hemoglobin F and genetic
testing. All subjects were transfused every 3₋4 weeks with packed red
cell since early years of life in an attempt to keep their
pretransfusion hemoglobin above 9.5 g/dl.
For each patient were
specified demographic and clinical data (family history, age, sex,
origin, consanguinity, age at diagnosis, age at the first blood
transfusion, anthropometric parameters); transfusion requirements and
complications related to secondary hemochromatosis; chelating therapy
(date of onset, type of chelation, modalities).
The size was
taken using the DETECTO metal gauge. The target size was calculated as
the average of the parents' heights plus 6.5 cm for boys or minus 6.5
cm for girls. Adult height was considered to be attained when growth
during the preceding year was less than 1 cm, with a bone age of over
15 years. Pubertal stages were assessed according to Tanner and
Marshall.[4] Arrested puberty is characterized by a lack of pubertal
progression over a year or more. Short stature is defined as height
less than two standard deviations (SDs) below the mean for age and
gender.[3] Body mass index (BMI) was calculated as weight (kg) divided by
the square of the height (m2) using reference charts for boys and
girls. Blood glucose was determined using the glucose oxidase method on
a Beckman Glucose Analyzer. Carbohydrate metabolism disorders were
assessed according to the American Diabetes Association (ADA).[5]
All
patients underwent hormonal evaluation testing including somatotropic,
gonadotropic, corticotropic, thyrotropic and parathyroid glands.
Evaluation of the GH/IGF-1 axis was performed by GH stimulation tests
as well as the insulin-like growth factor (IGF-1) and insulin-like
growth factor-binding-protein (IGFBP) concentrations compared to norms
for age and sex.[6] The diagnosis of GH deficiency was established on
an insufficient peak (less than 20 mIU /L) in response to two separate
pharmacological stimuli (insulin tolerance and glucagon-propranolol
tests). Subjects were arbitrarily classified according to GH peak in
partial GH deficiency (GH peak between 10 and 20 mIU /L) and a total
deficit if the values are less than 10 mIU/L. All subjects underwent a
basal cortisolemia and after intramuscular injection of
adrenocorticotropic hormone (Synacthen® test: 250 μg). An abnormal
response (a serum cortisol peak below 550 nmol/L or an increment of
less than 200 nmol/L from baseline or both) identifies adrenal
insufficiency. Thyroid function was assessed by measuring free
thyroxine (FT4) and thyrotrophic hormone (TSH). Subclinical
hypothyroidism is defined as a combination of high TSH (≥ 5 mIU/L) with
normal FT4 levels.
Skeletal age was evaluated according to
Greulich-Pyle atlas.[7] Bone mineral density (BMD) was performed by
Dual-energy x-ray absorptiometry (DXA) on L1-L4 lumbar spine and total
hips. Low bone mass was defined as Z-score values of –2.0 SDs or lower.
Iron overload was assessed using the mean serum ferritin levels and the MRI T2*. Cardiac and liver T2* were assessed by a validated technique based on MRI relaxometry at 1.5 T. For the heart T2*
images, all patients underwent a single breath hold multiecho bright
blood sequence with variable echo times (TEs). For the liver, a single
axial slice was obtained in the center of the organ using a multiecho
sequence, and a single breath hold was used to obtain images with the
same parameters. Excel spreadsheet was used for image analysis and
measurement of T2*. Images were
imported into a software for the region of interest (ROI) drawing. For
the heart, signal intensity was obtained using an ROI drawn through the
full thickness of the septum wall of the myocardial short axis image.
For the liver, the signal intensity was also provided using an ROI
covering the right lobe of the liver parenchyma and avoiding major
vessels. The same ROI was copied across all images for each organ. Each
image generated the values of both signal intensity (SI) and TEs which
were manually inputted into an Excel spreadsheet. The mean signal
intensity in each slice with varying TEs was used to fit the T2*
curve using the formula SI = Ke–TE/T2* in the spreadsheet. A
curve-fitting truncation model consisting of a monoexponential decay
curve with a linear fit was applied. Excel was applied as previously
described.[8] Myocardial ion concentration (MIC) was evacuated using
Carpenter curves.[9] Liver iron concentration (LIC) was calculated using
Hankins curves.[10] Values of cardiac T2*
(CT2*) <20 ms were considered to indicate cardiac siderosis which
was classified as moderate (10 ms< CT2* <20 ms) and severe (CT2*
<10 ms).[9] LIC >3 mg/g dry weight (dw) was considered to
indicate liver siderosis which was classified on mild (3 < LIC <7
mg/g dw). Moderate (7< LIC <15 mg/g dw) and severe (LIC >15
mg/g dw).[11] Serum ferritin (SF) concentration was measured every 3
months using standard enzyme immunoassay. The 12-month mean SF value
was considered.
The iron chelating treatments used were
subcutaneous deferoxamine (Desferal®), administered in two repeated
doses (40 mg/kg/day) 5-days-per-week, and oral chelators namely
deferasirox in a single dose (20 - 40 mg/kg/day) and deferiprone
in 3 daily taken (75 - 100 mg/kg/day). The combined treatment consisted
of combining deferoxamine with oral iron chelation. A group of 13
healthy subjects was used as control.
Written informed consent was obtained from the patients or their parents.
Statistical
analysis: All statistical procedures were performed using SPSS version
18.0. Results are presented in mean ± SDs. Pearson correlation analysis
and unpaired T student`s test were used. P value < 0.05 was
considered statistically significant.
Results
The
most recurrent mutation (SNP) single-nucleotide polymorphism) found was
Cd 39 (C > T) in 42%, IVS-I-110 (G>A) in 33%, Cd 6 (A>T) in
8%, and Cd 30 (G>C) in 4% of patients.
Growth:
sixteen (57%) of the studied patients had growth velocity standard
deviation score less than -2SDs. Among them, thirteen had a pubertal
delay. Bone maturation delay was present in all cases. Bone age delay
and chronological age were over one year in all patients. In the
absence of growth hormone deficiency, height changes with sex and age
are illustrated in figure 1.
Curves show that years, most children have a normal growth pattern up
to the age of 9 years, and a reduced or absent height gain during
puberty, which is more marked in boys than in girls. Circulating IGF-1
levels were significantly lower than controls (p = 0.00) (Figure 2).
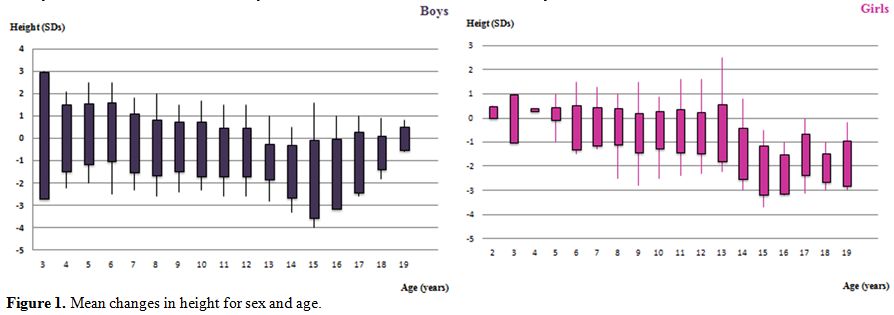 |
Figure 1.
Mean changes in height for sex and age. |
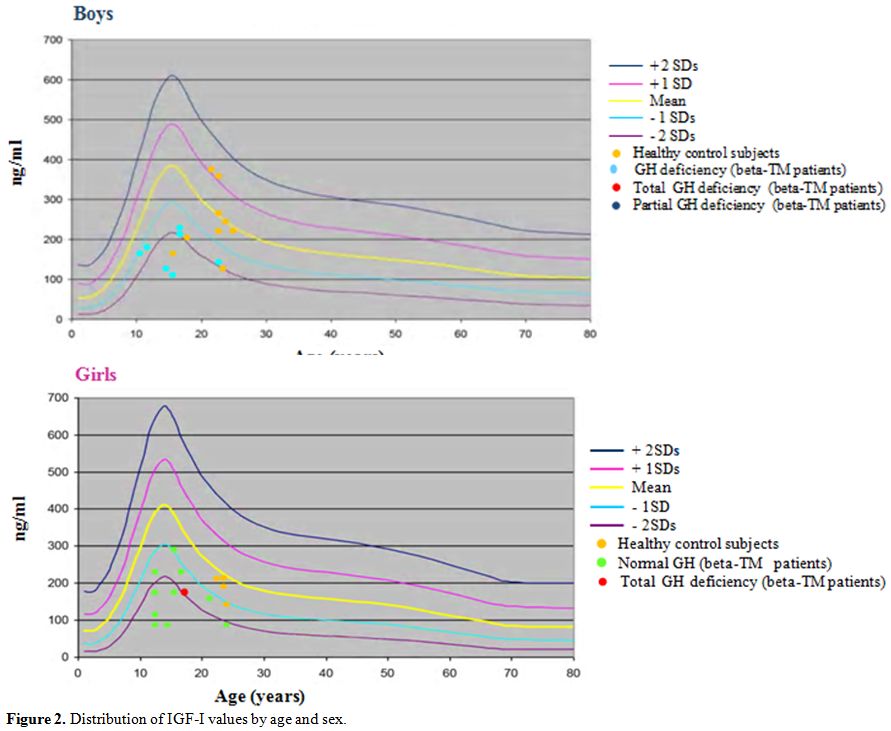 |
Figure 2. Distribution of IGF-I values by age and sex. |
Somatotropic function:
GH provocation tests showed an average peak GH levels in 18 patients
(40.22 ± 21.7 m IU/ L; range (21₋129). Ten patients (35%) had GH
deficiency, among them a partial GH deficiency was found in 5 cases
(16.14 mIU /L ± 2.87; range 3.7₋ 6.7) and a severe GH deficiency was
demonstrated in 5 other cases (5.16 mIU / L ± 1.35; range 13.7 ₋ 19.7).
Growth assessment for patients with complete GH deficiency showed that
of the 3 patients who had attained the adult age the parental target
size was reached in only one case. In patients with partial GH
deficiency, statural growth was normal in 3 cases. Only one patient had
a complete GH deficiency in contrast to normal IGF-1 level. Risk
factors, probably related to the occurrence of GH deficiency, are: a
history of splenectomy (p = 0.000), association with adrenal
insufficiency (p = 0.042), low bone mass (p = 0.037). The levels of
IGF-1 and IGFBP were significantly lower in patients with GH deficiency
than those without GH deficiency (p: 0.008 and 0.037 respectively).
Puberty:
spontaneous onset of puberty was obtained in 16 cases (9 boys and 7
girls) at a mean age of 15 years for boys (range 14-16 years) and 13
years for girls (range 11-15 years). Adult height was reached in 7
cases (5 boys and 2 girls) at a mean age of 20 years for boys (range
17-22 years) and 17 years for girls. Twelve of the studied patients
(42%) had a delayed puberty and hypogonadism requiring lifelong hormone
replacement therapy. Lack of pubertal progression was observed in 4
cases (2 girls and 2 boys), the absence of the onset of pubertal
development in 6 cases (5 boys and one girl) and a primary amenorrhea
in two other cases. Factors associated with pubertal disorders are
transfusion requirements before chelating therapy (p = 0.042) and
myocardial iron assessment by MRI T2* (p = 0.037).
Carbohydrate metabolism:
seventeen of the studied patients (60%) had disturbances of glucose
homeostasis with an impaired fasting glycemia in four cases, impaired
glucose tolerance in eight and diabetes mellitus in five. All of them
except one had been diagnosed after the age of 10. Diabetes was
preceded by a pre-diabetic stage in all cases with an average of 7
years (1 - 8). The mean age at the time of diagnosis was 20 ± 3.4 years
(12 - 15). No significant difference was seen between males and females
in the prevalence of diabetes mellitus. All diabetic patients had a
family history of type I or type II diabetes in their siblings, parents
or grandparents. The mean body mass index of diabetic patients was 19 ±
2.41 kg / m² (16.24-25). Overweight was noted in one patient. Two cases
had first presented with diabetic ketoacidosis. Islet cell antibodies,
insulin autoantibodies, and anti-glutamate decarboxylase were negative
in all cases. Sixteen patients were splenectomized. Serum ferritin
level in β-TM patients with diabetes and those without a history of diabetes were not significantly different. Severe cardiac loading (CT2*
≤10ms) was present in seven patients, and 10 patients had a severe iron
deposition in the liver (LIC >15 mg/g dw). All patients had
metformin as an antidiabetic agent associated with combined intensive
iron chelation therapy during a mean follow-up of 3 years (1-6 years).
Risk factors of carbohydrate metabolism disorders were: age at onset of
chelation therapy (p = 0.025) and ferritinemia; in fact, patients with
carbohydrate metabolism disorders had a higher average ferritin level
than those who did not, and the difference was statistically
significant (p = 0.03).
Thyroid function:
subclinical hypothyroidism was found in six patients (mean age 17 ±
3.14 years range 14 – 16) (mean TSH levels 5.97 ± 1.31 m IU /L range
5-9). Antithyroid antibodies were negative in all cases. Mean serum
ferritin level was 1405.8 ± 441.93 µg / l. Severe cardiac loading (CT2*
≤10ms) were seen in 3 cases, and severe liver loading (LIC >15 mg/g
dw) was observed in 4 cases. Only one patient required hormone
replacement therapy with levothyroxine. In all other cases, combined
chelation therapy allowed the normalization of thyroid hormone levels.
Adrenal function:
the cortisol peak was normal in 19 patients after ACTH stimulation.
Nine patients (32%) had adrenal insufficiency with a mean cortisol peak
of 413.93 ± 82.76 nmol / L(range 309 - 463). No patient was
symptomatic. A polyendocrinopathy was found in all cases. All patients
had combined chelation therapy with deferoxamine and deferasirox or
deferiprone. Splenectomy was performed in all cases. None of them had
received corticosteroid replacement therapy. Mean serum ferritin was
1,391.88 ± 661µg / l. Severe hepatic and cardiac loading was present in
three patients. Factors associated with the occurrence of adrenal
insufficiency were age (p = 0.033), history of splenectomy (p = 0.031),
number of transfusions received (p = 0.034), and associated GH
deficiency (p = 0.042).
Phosphocalcic metabolism and low bone mass:
out of 28 patients, only one girl was found to have hypoparathyroidism.
The mean age at diagnosis was 17 years. The patient initially
complained of extremity paresthesias, mean serum calcium was 1.6 mmol/L
(range 1.7 -1.9). Serum parathyroid hormone (PTH) level was low: 2 pg /
ml (normal: 12-72). Among the 28 studied patients, 16 had decreased
bone mineral density. Low bone mass was found in ten patients
predominantly male (7 patients). All patients had one or more
associated endocrinopathy. Factors related to the development of low
bone mass include: hypothyroidism (p = 0.007), GH deficiency (p =
0.037), decreased IGF-1 levels (p = 0.002), and iron overload on
cardiac (p = 0.021) and liver (p = 0.002) T2* MRI.
Associated endocrinopathies:
among the studied patients five had no endocrine disorder, and 23 (82%)
had at least one endocrinopathy with one endocrinopathy in six cases,
two in eight cases, three in four cases and four in five cases. The
mean age of these patients was 18.4 ± 1.3 years. The prevalence rates
of endocrine disorders are shown in Table 1.
There was a significant difference between mean serum ferritin in
thalassemic patients with endocrine complications (1660 ± 1208 μg /l)
and those without endocrinopathies (1166 ± 823 μg/l): p = 0.01.
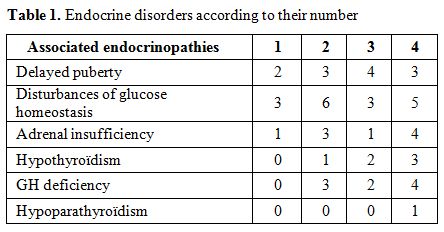 |
Table 1. Endocrine disorders according to their number |
Discussion
Endocrine abnormalities are widespread among multi‑transfused β
-TM patients. The most frequent endocrine complications reported are
growth retardation, delayed puberty, hypogonadism, carbohydrate
metabolism disorders, impaired thyroid, parathyroid and adrenal
functions. These complications are largely explained by the toxic
effect of iron overload secondary to chronic blood transfusions because
the human body lacks a mechanism to excrete excess iron.[12] The pathogenesis of the statural delay in β-TM patients is multifactorial.[13] In our study, in agreement with the findings of Skordis et al.,[14]
growth disorders varied depending on age at presentation. In early
childhood, growth retardation is mainly due to hypoxia, anemia,
ineffective erythropoiesis and nutritional factors. During late
childhood, iron overload affecting GH-IGF-1 axis and other potential
endocrine complications are the main factors affecting growth. After 11
years of age, delayed or arrested puberty is an important contributing
factor to growth failure. In accordance with other studies, maintaining
a pretransfusion hemoglobin level above 9–10.5 g/ dl promotes normal
growth during the first years of life for the majority of patients in
our study. Total growth hormone deficiency is an associated factor that
may contribute to the growth delay in patients with β-thalassemia
major. In our study, we found that five out of 28 had complete growth
hormone deficiency. Four of them had a height 2 standard deviations
below the mean. On the other hand, among the five patients with partial
growth hormone deficiency, four had normal growth rate suggesting the
likely secondary origin of growth hormone deficiency in β-thalassemia
major patients. Several studies assessed the GHRH-GH-IGF-1axis function
pointing out its impairment in a large number of short patients.
Neurosecretory GH disorders with different prevalence are reported in
thalassemia patients with short stature while contradictory data are
available on GH reserve. It was reported normal or reduced with a wide
variability (8-80%) in short patients, due to defects in the pituitary
gland and/or in the hypothalamus.[15]. Other authors
have reported normal GH and GHBP levels but low levels of IGF-1 and
IGFBP which are not properly increased with IGF-1 generation test,
suggesting that insensitivity to GH action may be the cause of abnormal
growth.[16] The reduction of serum IGF-1 levels in β-TM with no growth hormone deficiency in the different stimulation tests supports these findings.[17,18] However, a lack of correlation between IGF-1, IGFBP, and height SDS in β-TM
children with growth failure may indicate that growth failure is not
specifically related to GH-IGF-1 axis. In accordance with what has been
described, we have found IGF-1 levels to be significantly lower
compared to controls without any growth hormone deficiency. Also,
patients with growth hormone deficiency had lower levels of IGF-1 than
non-deficient patients. Hypogonadotropic hypogonadism, is the most
frequent endocrinopathy in patients with transfusion-dependent
thalassemia.[19,20] In male patients, clinical
presentations of hypogonadotropic hypogonadism include lack, delay,
and/or block of pubertal sexual maturation and, in adult life,
decreased libido, erectile dysfunction, worsened sense of well-being,
and lower quality of life. Spermatogenesis is impaired, and the volume
of ejaculate is decreased. In female patients, hypogonadism is
clinically diagnosed by the absence of pubertal development or
discontinuation or regression of the maturation of secondary sex
characteristics.[20] Low serum concentrations of sex
hormones and gonadotropins confirm the diagnosis. Several questions
were asked about their diagnosis, their central or peripheral origin
and their reversibility to the intensification of the iron chelating
treatment. The GnRH (gonadotrophin-releasing hormone) test is unhelpful
in the clinical assessment of the hypothalamic-pituitary axis in β-TM
patients especially when the chronological and bone ages have not
reached pubertal levels. The ability of the testes to produce
testosterone under the stimulatory effect of chorionic gonadotropins
reflecting the hypophyseal origin of hypogonadism. In girls, pubertal
anomalies are mainly represented by menstrual cycle disorders.
According to a study by Borgna-Pignatti et al.,[21] studying 118 patients with β-TM,
only 23 (19.4%) had spontaneous menarche. Our study has shown that
nearly two-thirds of patients have carbohydrate metabolism disorders.
According to the ADA criteria, 12 patients (42%) had a pre-diabetic
state with impaired glucose tolerance and impaired fasting glucose,
while five patients had developed diabetes. However, none of them
required insulin therapy. The incidence of pre-diabetic state is
considerably higher than the 4.2% of patients developing IDDM in a
recent study reported by Bejaoui et al. in a Tunisian multicenter
study.[22] The higher prevalence in our study could
be related to the age of patients being studied, with lower rates in
younger patients. Indeed, according to the French national register,
including 215 β-TM, diabetes was found in 13 patients (6%).[23]
According to the same register, it rose from 1.3% before the age of 15
to 4.1% between 15 and 24 years, reaching more than 11% in those above
the age of 25 years. The pathogenesis of diabetes in β-TM
patients is not fully understood. Studies report the early occurrence
of insulin resistance and hyperinsulinemia leading to IGT.[24-27]
With advancing age, the persistence of insulin resistance exhausts
β-cells and reduces insulin secretion leading to DM. However, other
studies report that a defective insulin secretion resulting from toxic
effects of iron deposition in the pancreas may precede the development
of glucose intolerance. As described before, myocardial and hepatic T2*
values were significantly higher in patients with carbohydrate
metabolism disorders. Diabetes is often associated with other
endocrinopathies. Among the studied patients, six (21%) had primary
hypothyroidism (3 girls and 3 boys) with a mean age of 17 ± 3.14 years.
Thyroid dysfunction has been reported in 13–60% of patients with
thalassemia, but its severity is variable in different series. These
discrepancies cannot be attributed to differences in patients' ages,
but rather to difference treatment protocols, including differing
transfusion rates and chelation therapies.[28] Milder forms of thyroid dysfunction are much more common.[29-31] Subclinical hypothyroidism was found in all cases with thyroid levels mean of 5.97 ± 1.31 m IU / L. According to others,[32]
antithyroid antibodies were negative in all cases. Mean serum ferritin
level was 1405.8 ± 441.93 ng / ml. Severe overload was shown in 3 cases
on Cardiac T2* analysis, and liver
overload in 4 cases. Only one patient required hormone replacement
therapy with levothyroxine. In all other cases, combined chelation
treatment allowed the normalization of thyroid hormone levels. In a
large prospective study, hypogonadism was diagnosed in 86% of patients,
hypoparathyroidism in 23% of patients, and hypothyroidism in 18% of
cases. Among 5 diabetic studied patients , pubertal development delay
was found in 4 cases, GH deficiency in two cases and subclinical
hypothyroidism in 2 cases. Adrenal insufficiency is not a rare
complication in β-TM.
However, it is of little or no clinical impact under basal conditions.
Accordingly, glucocorticoid replacement therapy might be advised only
for stressful conditions.[33] HPT has been considered
as a typical complication of the second decade of life in
transfusion-dependent patients with TM. It is thought to be mainly the
consequence of iron deposition in the parathyroid glands. In most
patients the onset of HPT was preceded or followed by other endocrine
complications.[34] Our patient with
hypoparathyroidism had a total absence of pubertal development,
diabetes and low bone mass. Low bone mass represent prominent causes of
morbidity in young adults of both genders with β-TM and the incidence of low bone mass in well treated β-TM
patients has been found to be approximately 40–50% .In addition to
genetic factors, bone marrow expansion, direct iron toxicity to
osteoblasts, deferoxamine iron chelation, and endocrine gland
involvement appear to play a major role. According to others, bone
mineral density was significantly lower in β-TM patients with endocrinopathy. Subclinical impairment of adrenocortical function in patients with β-TM
is not uncommon, however, it is of little or no clinical impact under
basal conditions but may have a potential relevance during stressful
events. Accordingly, glucocorticoid treatment coverage might be advised
only for stressful conditions. Clinical adrenal insufficiency and
adrenal crisis are very rare. Parathyroid insufficiency is a rare and
late complication of iron overload.[35] The majority
of patients is asymptomatic or has moderate impairment, thus requiring
a periodic follow-up of the phosphocalcic balance in β-TM
patients. This complication is often associated with other
endocrinopathies that they always precede. Our patient with
hypoparathyroidism had a total absence of pubertal development ,
diabetes and low bone mass. With longer life expectancy, low bone mass
becomes increasingly marked. Our study revealed that 35% of the
patients had low bone mass. According to others, we found the bone
mineral density is significantly lower in patients with associated
endocrinopathy. Our study showed higher prevalence of multiple
endocrine complications than that recently reported by De Sanctus et
al.,[32] however the percentage of patients without multiple endocrine complications are similar in the two study groups.
Conclusions
Endocrine and metabolic disorders are very common among multi-transfused β-TM
patients. Ferritin concentrations are not a reliable predictor of these
complications. Patients with evidence of cardiac iron overload have
more frequently endocrinopathies. This suggests that tissue iron
loading is a crucial factor leading to these disorders. Early detection
of these abnormalities as well as multidisciplinary management with
standardized protocols are the best means to ensure a better quality of
life for patients. Moreover, the promise of new chelators in
development can be viewed with an intelligible optimism for a new age
of iron chelation therapy.
.
References
- Telfer PT, Warburton F, Christou S,
Hadjigavriel M, Sitarou M, Kolnagou A, Angastiniotis
M. Improved survival in thalassemia major patients on switching from
desferrioxamine to combined chelation therapy with desferrioxamine and
deferiprone. Haematologica 2009; 94 (12):1777-1778. http://dx.doi.org/10.3324/haematol.2009.009118 PMid: 19815834

- De
Sanctis V, Roos M, Gasser T, Fortini M, Raiola G, Galati MC. Italian
Working Group on Endocrine Complications in Non-Endocrine Diseases.
Impact of long-term iron chelation therapy on growth and endocrine
functions in thalassaemia. J Pediatr Endocrinol Metab 2006;
19(4):471-480. PMID:16759032
- Sempé. Sempé M, Pédron G, Roy-Pernot MP. Auxologie méthode et séquences. Théraplix. Paris; 1979.
- Tanner J.M. Growth at adolescence. 2nd Edition Blackwell Scientific Publications; 1962.
- Diagnosis and classification of diabetes mellitus. American diabetes association. Diabetes Care 2010; 33: 62-69. http://dx.doi.org/10.2337/dc10-S062 PMid: 20042775
- Brabant
G, von zur Mühlen A, Wüster C, Ranke MB, Kratzsch J, Kiess W,
Ketelslegers JM, Wilhelmsen L, Hulthén L, Saller B, Mattsson A, Wilde
J, Schemer R, Kann P. German KIMS Board. Serum insulin-like growth
factor I reference values for an automated chemiluminescence
immunoassay system: results from a multicenter study. Horm Res 2003;
60:53-60. http://dx.doi.org/10.1159/000071871 PMid:12876414
- Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand and wrist. 2nd Ed. Stanford University Press; 1959.
- Fernandes
JL, Sampaio EF, Verissimo M, Pereira FB, da Silva JA, de Figueiredo GS,
Kalaf JM, Coelho OR . Heart and liver T2 assessment for iron overload
using different software programs. Eur Radiol 2011; 21(12):2503–2510. http://dx.doi.org/10.1007/s00330-011-2208-1 PMid:21842212
- Carpenter
JP, He T, Kirk P, Roughton M, Anderson LJ, de Noronha SV, Sheppard MN,
Porter JB, Walker JM, Wood JC, Galanello R, Forni G, Catani G, Matta G,
Fucharoen S, Fleming A, House MJ, Black G, Firmin DN, St Pierre TG,
Pennell DJ. On T2* magnetic resonance and cardiac iron. Circulation
2011; 123(14):1519–1528. http://dx.doi.org/10.1161/CIRCULATIONAHA.110.007641 PMid:21444881
- Hankins
JS, Mccarville MB, Loeffler RB et al. R2* magnetic resonance imaging of
the liver in patients with iron overload. Blood 2009;
113(20):4853–4855. http://dx.doi.org/10.1182/blood-2008-12-191643 PMid:19264677
- Di
Tucci AA, Matta G, Deplano S, Gabbas A, Depau C, Derudas D, Caocci G,
Agus A, Angelucci E. Myocardial iron overload assessment by T2*
magnetic resonance imaging in adult transfusion dependent patients with
acquired anemias. Haematologica 200 ; 93(9):1385–1388. http://dx.doi.org/10.3324/haematol.12759 PMid:18603557
- Ouederni
M , Ben Khaled M , Mellouli F , Ben Fraj E , Dhouib N , Yakoub IB ,
Abbes S , Mnif N , Bejaoui M .Myocardial and liver iron overload,
assessed using T2* magnetic resonance imaging with an excel spreadsheet
for post processing in Tunisian thalassemia major patients. Ann Hematol
2017; 96(1):133-139. http://dx.doi.org/10.1007/s 00277-016-2841-5 PMid:27730342
- Soliman A T, Khalafallah H, Ashour R. Growth and factors affecting it in thalassemia major. Hemoglobin 2009; 33(S1):S116–S126. http://dx.doi.org/10.3109/03630260903347781 PMid:20001614
- Skordis
N, Kyriakou A. The multifactorial origin of growth failure in
thalassaemia. Pediatr Endocrinol Rev 2011; 8 Suppl 2:271-277.
PMid:21705977
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis 2010;5:11. http://dx.doi/10.1186/1750-1172-5-11 PMID: 20492708
- Kyriakou
A, Skordis N.Thalassaemia and Aberrations of Growth and Puberty.
Mediterr J Hematol Infect Dis. 2009; 1(1): e2009003. PMID: 21415985
- De
Sanctis V, Skordis N, Galati MC, Raiola G, Giovannini M, Candini G,
Kaffe K, Savvides I, Christou S . Growth hormone and adrenal response
to intramuscular glucagon test and its relationship to IGF-1 production
and left ventricular ejection fraction in adult B-thalassemia major
patients. Pediatr Endocrinol Rev 2011; 8 Suppl 2:290-294. PMid:21705980
- Pincelli
AI, Masera N, Tavecchia L, Perotti M, Perra S, Mariani R, Piperno A,
Mancia G, Grassi G, Masera G. GH deficiency in adult B-thalassemia
major patients and its relationship with IGF-1 production. Pediatr
Endocrinol Rev 2011; 8 Suppl 2:284-289. PMid:21705979
- De
Sanctis V, Elsedfy H, Soliman AT, Elhakim IZ, Pepe A, Kattamis C,
Soliman NA, Elalaily R, El Kholy M, Yassin M. Acquired hypogonadotropic
hypogonadism (AHH) in thalassaemia major patients: An underdiagnosed
condition? Mediterr J Hematol Infect Dis 2016, 8(1): e2016001. http://dx.doi.org/10.4084/MJHID.2016.001
- De
Sanctis V, Soliman AT, Elsedfy H, Albu A, Al Jaouni S, Anastasi S,
Bisconte MG, Canatan D, Christou S, Daar S, Di Maio S, El Kholy M,
Khater D, Elshinawy M, Kilinc Y, Mattei R, Mosli HH, Quota A, Roberti
MG, Sobti P, AL Yaarubi S, Canpisi S, Kattamis C. Review and
recommendations on management of adult female thalassemia patients with
hypogonadism based on literature review and experience of ICET-A
network specialists. Mediterr J Hematol Infect Dis 2017, 9(1):
e2017001. http://dx.doi.org/10.4084/MJHID.2017.001
- Borgna-Pignatti
C, De Stefano Ρ, Zonta Μ, Vullo C, De Sanctis V, Melevendi C, Naselle
A, Masera G, Terzoli S, Gabitti V, Piga A.Growth and sexual maturation
in thalassemia major. J Pediatr 1985 ; 106: 150-156. PMid:3965675
- Bejaoui
M, Guirat N. Beta thalassemia major in a developing country:
epidemiological, clinical and evolutionary aspects. Mediterr J Hematol
Infect Dis 2013;5(1):e2013002. http://dx.doi.org/10.4084/MJHID.2013.002 PMid:23350015
- Thuret
I, Pondarré C, Loundou A, Steschenko D, Girot R, Bachir D, Rose C,
Barlogis V, Donadieu J, De Montalembert M, Hagege I, Pegourie B, Berger
C, Micheau M, Bernaudin F, Leblanc T, Lutz L, Galactéros F, Siméoni MC,
Badens C. Complications and treatment of patients with β-thalassemia in
France: results of the National Registry. Haematologica 2010;
95(5):724-729. http://dx.doi.org/10.3324/haematol.2009.018051 PMid: 20007138
- Toumba
M, Sergis A, Kanaris C, Skordis N . Endocrine complications in patients
with thalassaemia major. Pediatr Endocrinol Rev 2007; 5: 642-648.
PMId:18084158
- Gamberini
MR, Fortini M, De Sanctis V, Gilli G, Testa MR.. Diabetes mellitus and
impaired glucose tolerance in thalassaemia major: incidence,
prevalence, risk factors and survival in patients followed in the
Ferrara Center.Pediatr Endocrinol Rev 2004 ; 2 (Suppl 2): 285-291.
PMid:16462713
- Au
WY, Lam WW, Chu W, Tam S, Wong WK, Liang R, Ha SY . A T2* magnetic
resonance imaging study of pancreatic iron overload in thalassemia
major. Haematologica 2008; 93:116-119. http://dx.doi.org/10.3324/haematol.11768 PMid:18166794
- Bas
M, Gumruk F, Gonc N, et al. Biochemical markers of glucose metabolism
may be used to estimate the degree and progression of iron overload in
the liver and pancreas of patients with β-thalassemia major. Ann
Hematol 2015;94:1099-2104. http://dx.doi.org/10.1007/s00277-015-2342-y PMid:25740381
- Phenekos
C, Karamerou A, Pipis P, Constantoulakis M, Lasaridis J, Detsi S,
Politou K. Thyroid function in patients with homozygous β-thalassemia.
Clin Endocrinol (Oxf) 1984; 20:445–450. PMid:6424976
- Magro
S, Puzzanio P, Consarino C, Galati MC, Morgione S, Porcelli D, Grimaldi
S, Tancre D, Arcuri V, De Santis V. Hypothyroidism in patients with
thalassemia syndromes. Acta Haematol (Basel) 1990; 84:72–76. http://dx.doi.org/10.1159/000205032 PMid:2120889
- Depaz
G, Deville A, Coussement N, Manassero J, Mariani R. Thyroid function in
thalassemia major. Ann Pediatr (Paris) 1985 ; 32:809–811. PMid:4091451
- Landau
H, Matoth I, Landau-Cordova Z, Goldfarb A, Rachmilewitz EA, Glaser B.
Cross-sectional and longitudinal study of the pituitary-thyroid axis in
patients with thalassaemia major. Clin Endocrinol (Oxf) 1993 ;
38:55–61.PMid:8435886
- De
Sanctis V, Elsedfy H , Soliman AT., Elhakim IZ
, Kattamis C, Soliman NA, Elalaily R. Clinical and
Biochemical Data of Adult Thalassemia Major patients (TM) with Multiple
Endocrine Complications (MEC) versus TM Patients with Normal Endocrine
Functions: A long-term Retrospective Study (40 years) in a Tertiary
Care Center in Italy. Mediterr J Hematol Infect Dis 2016; 8(1):
e2016022. http://dx.doi.org/10.4084/MJHID.2016.022 PMid: 27158435
- Huang
KE , Mittelman SD, Coates TD, Geffner ME, Wood JC. A significant
proportion of thalassemia major patients have adrenal
insufficiencydetectable on provocative testing. J Pediatr Hematol
Oncol. 2015; 37(1):54-59. http://dx.doi.org/10.1097/MPH.0000000000000199 PMid:24942024
- De
Sanctis V , Soliman AT, Canatan D , Elsedfy H, Karimi M , Daar S ,
Rimawi H , Christou S, Skordis N , Tzoulis P, Sobti P, Kakkar S,
KilincY, Khater D, Alyaarubi SA, Kaleva V, Lum SH, Yassin MA, Saki F,
Obiedat M, Anastasi S, Galati MC, Raiola G, Campisi S , Soliman N,
Elshinawy M, Al Jaouni S, Di Maio S, Wali Y, Elhakim IZ, Kattamis C. An
ICET- A survey on Hypoparathyroidism in Patients with Thalassaemia
Majorand Intermedia: A preliminary report. Acta Biomed 2018;
16;88(4):435-444. http://dx.doi.org/10.23750/abm.v88i4.6837 PMid:29350657
- Casale
M, Citarella S, Filosa A, De Michele E, Palmieri F, Ragozzino A,
Amendola G, Pugliese U, Tartaglione I, Della Rocca F, Cinque P, Nobili
B, Perrotta S. Endocrine function and bone disease during long-term
chelation therapy with deferasirox in patients with β-thalassemia
major. Am J Hematol 2014; 89(12):1102-1106. http://dx.doi.org/10.1002/ajh.23844 PMid:25197009
[TOP]