Mohammad Ehsan Jaripour1#, Kourosh Hayatigolkhatmi1#, Vahid Iranmanesh1, Farhad Khadivi Zand1, Zahra Badiei2, Hamid Farhangi2, Ali Ghasemi2, Abdollah Banihashem2, Reza Jafarzadeh Esfehani3 and Ariane Sadr-Nabavi1,3,4*.
1 Iranian Academic Center for Education, Culture and Research, (ACECR), Mashhad, Iran.
2 Department of Pediatric Diseases, School of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran.
3 Department of Medical Genetics, School of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran.
4 Medical Genetics Research Center, School of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran.
# These authors contributed equally to this work.
Corresponding
author: Ariane Sadr-Nabavi. Assistant professor in medical genetics and
head of the ACECR medical genetics center of Mashhad. Medical Genetics
Department, School of Medicine, Mashhad University of Medical Sciences,
Azadi Square, Mashhad, Iran. Tel: 00985138002226. Email:
Sadrnabavia@mums.ac.ir
Published: July 1, 2018
Received: March 13, 2018
Accepted: June 14, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018042 DOI
10.4084/MJHID.2018.042
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background and Objective: ß-thalassemia results from a diverse range of mutations inside the hemoglobin subunit β (HBB)
gene. In a study of β-thalassemia carriers and some of their at-risk
fetuses in the Khorasan province of Iran, we aimed to recognize the
most common mutations in the region. We also investigated a possible
link between these mutations and some of the relevant hematological
indices.
Methods: Amplification-refractory mutation system-PCR (ARMS-PCR) was used to detect the typical HBB mutations among 1593 individuals, suspected of having a mutated HBB allele from March/2011 to January/2018. Sanger sequencing of HBB
had been performed, where ARMS-PCR was uninformative. In some cases,
reverse dot blot was utilized. Analysis of variance was used to compare
parametric variables.
Results:
Among 1273 ß-thalassemia carriers, the prevalence of the mutations were
reported as follows: IVS-I-5 (42.03%), IVS-II-1 (11.23%), codons 8/9
(4.79%), codon 44 (4.56%), codon 15 (3.53%), Los Angeles (2.91%), codon
5 (2.75%), IVS-I-110 (2.51%), -88 (2.20%) and other mutations were less
than 2% of all of the reported mutations. 644 conceptions were
subjected to prenatal diagnosis, using chorionic villus sampling. 118
cases were reported as normal. 352 cases were detected as carriers. 174
cases were diagnosed as affected. There was a significant difference in
mean corpuscular volume and hemoglobin A2 levels between the nine most
commonly reported mutation types (p<0.001).
Conclusion: This study makes a reliable guide for ß-thalassemia diagnosis in the region. The possibility of a correlation between HBB mutations and hematological indices opens a gate of future investigations.
|
Introduction
ß-thalassemia
refers to a highly common hereditary hematological disorder, caused by
depletion or absence of the ß-globin synthesis. Regarding molecular
aspects, this autosomal recessive disorder occurs following mutations
which affect chromosome 11 in ß chain locus, responsible for coding a
146 amino acids polypeptide.[1,2] Diminution of ß
chain production, in turn, results in excess of α-globin chains,
accumulation of extra hemoglobin (Hb) inside the red blood cells
(RBCs), destruction of mature RBCs in spleen (hemolysis) and bone
marrow hyperplasia as a result of its inconclusive reparative efforts.[1,2]
The broad spectrum of mutations in the β-globin gene brings a variety
of phenotypically different features in patients, which enable
clinicians to classify these disorders into different types. These
subtypes are known as non-transfusion dependent (ND) and transfusion
dependent (TD) ß-thalassemia (AKA: minor ß-thalassemia and major
ß-thalassemia respectively).[1,2] Majority of these
mutations are nucleotide substitutions, frameshifts, and small
deletions. Large deletions are rarely involved in the development of
ß-thalassemia.[1-11] These mutations affect synthesis
of the ß-globin chain differently. Some of them bring a very mild
reduction in the ß-globin production (ß++ allele) while others may result in marked depletion (ß+ allele) or complete absence (ߺ allele) of the β-globin polypeptide.[1,2]
ß-thalassemia, as discussed, can be presented with various severities
and differences among patients’ hematological indices, based on the
responsible mutation type.[1-5]
Researches prove
that ß-thalassemia is one of the most frequent genetic diseases
worldwide, with a range of ethnically and geographically distributed
mutations.[1,2] Moreover, the large number of carriers
is a warning for the health system and an emphasis on the importance of
preventing programs.[3-14] ß-globin genetic mutations
distribute among every ethnic group. Identification of these mutations
helps authorities for more accurate evaluations and more practical
prevention programs.[3-14]
The purpose of this
study was to recognize the most common mutations related to
ß-thalassemia in the Khorasan province of Iran and to find the possible
relation of these mutations with some of the relevant hematological
indices. These indices are presented in complete blood count (CBC) and
Hb electrophoresis tests (identifying HbA1, HbA2, and HbF). These
indices include RBC, Hb, mean corpuscular volume (MCV), mean
corpuscular hemoglobin (MCH).
Material and Methods
The region of study:
Khorasan province of Iran is the largest province of the country. The
greater Khorasan consists of Razavi Khorasan, Northern Khorasan, and
Southern Khorasan. 8,000,000 people are estimated to reside in the
greater Khorasan. The study happened in the Razavi Khorasan with
~6,500,000 residents, within the academic center for education,
culture, and research (ACECR) medical genetics laboratory. The ACECR
laboratory has received most of the ß-thalassemia detection referrals
over the Razavi Khorasan province and even some districts of the
Northern and the Southern Khorasan. We estimate that the ACECR
laboratory is responsible for ß-thalassemia detection among at least
~70% of the Razavi Khorasan’s population, which is an estimation of
4,550,000 individuals. Estimations are based on the latest national
census program and details provided by the local health center through
personal communications.
Study subjects:
The population investigated in this study comprises of 1593 individuals
with Fars ethnicity, suspected of possessing a mutated allele for
ß-thalassemia and referred to the ACECR diagnostic medical genetics
laboratory for molecular diagnosis. Personal consent form to obtain the
permission to use the patients’ samples and the relevant data in the
research performed by the ACECR was signed by every single individual
of this study. The ACECR ethics committee, which functions under the
regulations of the national medical ethics committee, approved these
consent forms. Clinical criteria for suspicious ß-thalassemia was
determined by the hypochromic microcytic anemia, including decreased
MCV (<80 fL) and MCH (<27 pg/cell) and unusual findings in Hb
electrophoresis, including elevated HbA2 (≥ 3.5 %) or HbF (≥ 1%) and
other abnormally high Hb variants.[15] Peripheral
blood (PB) samples from subjects were collected in EDTA containing
tubes. Furthermore, chorionic villus samples (CVS) were collected from
644 conceptions, whose fetuses were at risk of inheriting two mutated
alleles for hemoglobin subunit β (HBB)
gene. Samples were collected by a neonatologist and sent to the ACECR
medical genetics laboratory for ß-thalassemia prenatal diagnosis (PND).
All samples (PB and CVS) were collected since March of 2011 up to
January of 2018.
DNA extraction: DNA from selected individuals was extracted using standard salting out method as described by.[16]
DNA from CVS was isolated using QiAmp® DNA Blood Mini Kit (Qiagen GmbH,
Hilden, Germany) as described by the company instructions.
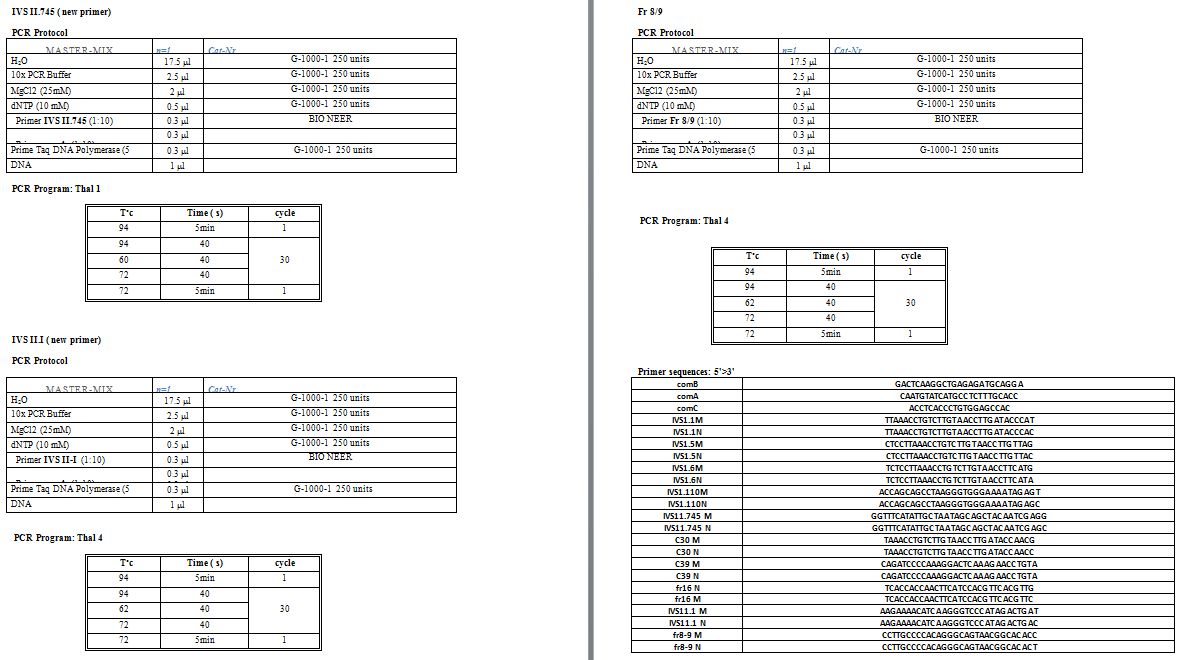
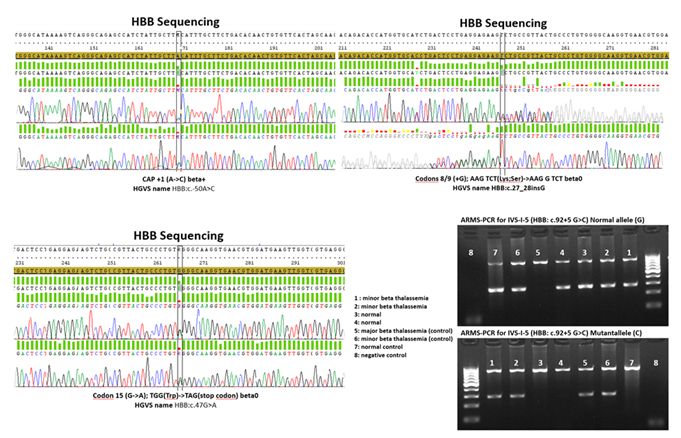
Mutation detection:
For each amplification-refractory mutation system (ARMS-PCR) was used
to detect ten common β-globin mutations: IVS-I-5, IVS-II-1, IVS-I-110,
IVS-I-1, IVS-II-745, IVS-I-6, codon 30, codon 39, codon 819, and codon
16.[17] In cases, where none of the mentioned variants was detected; standard Sanger sequencing of the HBB had been performed to reveal the mutation. Supplementary table
1 shows primer sequences, micro-tube components and the thermal
protocol used for the mentioned methods. Moreover, in some emergency
cases, reverse dot blot (strip assay) was conducted using Thalassemia
StripAssays® kit (ViennaLab, Vienna, Austria). For ß-thalassemia PND
cases, we have routinely confirmed the result with two alternative
molecular techniques. (See supplementary files)
Statistical analysis:
Data was analyzed using the statistical package for social sciences
(SPSS) version 22 (IBM Inc, Chicago, Il, USA). Continuous data were
checked for normality using the Kolmogorov-Smirnov test. Means and
standard deviations (SD) were used to describe continuous variables
while frequency and percentage were used to describe categorical
variables. Analysis of variance (ANOVA) was used to compare parametric
variables, including RBC count, Hb, MCV, MCH, HbA2, and HbA1, between
the mutation categories while the Kruskal-Wallis test was used to
compare non-parametric variable (HbF) values between mutation groups.
Multinomial logistic regression was performed to assess the
relationship between the mutation categories and the study parameters
considering other mutations than the nine evaluated (mostly reported
mutations) as the reference category. The odds ratio (OR) and 95%
confidence interval (CI) for OR were presented along with p-value for
the regression model. Values of p less than 0.05 were considered
statistically significant.
Results
As indicated in figure 1,
among 1273 ß-thalassemia carriers studied over seven years (out of 1593
suspected referrals, whom 320 of them were not confirmed to have a
pathogenic HBB variant based on our diagnostic molecular methods as
discussed in the ‘material and methods’ section) the most commonly
reported mutations were in a decreasing order of: IVS-I-5 (HBB: c.92+5G>C/A/T), IVS-II-1 (HBB: c.315+1G>A/C), codons 8/9 (HBB:c.27_28insG), codon 44 (HBB:c.135delC), codon 15 (HBB:c.48G>A), Los Angeles (HBB:c.364G>C), codon 5 (HBB:c.17_18delCT), IVS-I-110 (HBB:c.93-21G>A), -88 (HBB:c.-138C>A/G/T), IVS-I-6 (HBB:c.92+6T>C), codon 39 (HBB:c.118C>T), IVS-II-745 (HBB:c.316-106C>G), -29 (HBB:c.-79A>G), δβ (various large deletions), 25bp deletion (HBB:c.93-22_95del), HbS (HBB:c.20A>T) as all addressed by HbVar database.[18] The rest of the mutations were less than 1% all of the reported variants.
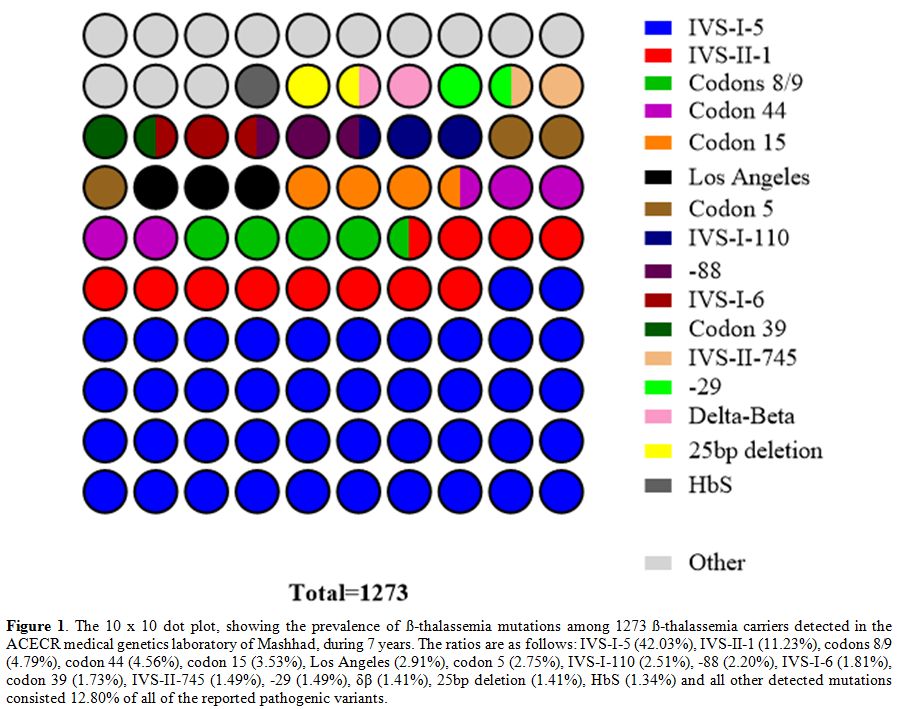 |
Figure 1. The 10 x 10 dot
plot, showing the prevalence of ß-thalassemia mutations among 1273
ß-thalassemia carriers detected in the ACECR medical genetics
laboratory of Mashhad, during 7 years. The ratios are as follows:
IVS-I-5 (42.03%), IVS-II-1 (11.23%), codons 8/9 (4.79%), codon 44
(4.56%), codon 15 (3.53%), Los Angeles (2.91%), codon 5 (2.75%),
IVS-I-110 (2.51%), -88 (2.20%), IVS-I-6 (1.81%), codon 39 (1.73%),
IVS-II-745 (1.49%), -29 (1.49%), δβ (1.41%), 25bp deletion (1.41%), HbS
(1.34%) and all other detected mutations consisted 12.80% of all of the
reported pathogenic variants.
|
During
the years of this study, 644 conception cases on the formerly mentioned
ß-thalassemia carrier couples were subjected to PND, using CVS samples
as described in the “methods” section.
For 118 cases which were
reported to carry no detectable pathogenic variant and 352 cases which
were detected as carriers (heterozygous with one pathogenic variant in HBB)
and expected to show the ND ß-thalassemia phenotype, pregnancies were
followed up till the delivery. Whereas 174 cases, which were diagnosed
as homozygous with two pathogenic variants in HBB
and expected as TD ß-thalassemia phenotype, have received the proper
genetic counseling based on the regional guidelines and prevention
programs.[13,14]
Further, the relation of the
first nine more commonly reported mutations with the hematological
indices was observed as mentioned previously. There was a significant
difference in MCV and HbA2 levels between the mutation types
(p<0.001) (Table 1).
Multinomial logistic regression model revealed that mutation
categorized as IVS-II-I was associated with increased risk for higher
MCH (p=0.01, OR=1.15, 95% CI for OR= 1.03 and 1.28) and HbA2 (p=0.002,
OR= 1.26, 95% CI for OR= 1.09, 1.46) and lower MCV (p<0.001,
OR=0.93, 95% CI for OR= 0.90, 0.97) compared to other mutations.
Furthermore, the codons 8/9 mutation was found to be associated with
significant increase in HbF values compared to other mutations (p=0.04,
OR= 1.05, 95% CI for OR= 1.00, 1.09).
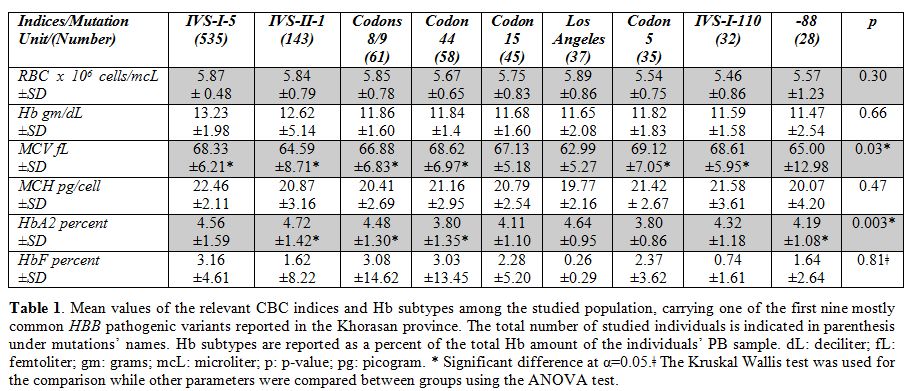 |
Table 1. Mean values
of the relevant CBC indices and Hb subtypes among the studied
population, carrying one of the first nine mostly common HBB
pathogenic variants reported in the Khorasan province. The total number
of studied individuals is indicated in parenthesis under mutations’
names. Hb subtypes are reported as a percent of the total Hb amount of
the individuals’ PB sample. dL: deciliter; fL: femtoliter; gm: grams;
mcL: microliter; p: p-value; pg: picogram. * Significant difference at
α=0.05.ǂ The Kruskal Wallis test was used for the comparison while
other parameters were compared between groups using the ANOVA test. |
Discussion
Despite
the spacious area and of course the massive population of the greater
Khorasan, this province shows one of the lowest prevalence for
ß-thalassemia in Iran.[6,7] Still, ß-thalassemia is one of the most hereditary hematological disorders reported routinely in this province.[6,7]
Prior to the current study, there was almost no similar report on the
ratio of ß-thalassemia mutations over this area, especially with such a
vast pool of carriers. Just a previous study has named the codons 8/9
variant as the most prevalent mutation in the region.[6]
Our research has contradicted this previous finding and identified the
IVS-I-5 as the most frequent mutation, confirming the review by Mahdieh
et al., 2016,[7] while the codons 8/9 is recognized as
the third most common mutation. The ß-thalassemia mutations’ prevalence
reported by this study can act as a reliable resource for molecular
diagnostic laboratories over the province, due to its considerable
number of studied cases and the long time-period of observation.
However, we have experienced some limitations, including the
unwillingness of some of the potential carrier couples to enroll in
genetic counseling sessions and undertaking the ß-thalassemia detection
tests following clinical referrals. Generally, since the two variants
of IVS-I-5 and IVS-II-1 consist over 50% of the total ß-thalassemia
causing mutations in the province, it will be of vital importance for
the local diagnostic laboratories to initiate their molecular diagnosis
with the specific focus on these two variants.
Additionally, we
investigated the relation of the first nine most commonly reported
mutations with the hematological indices as mentioned formerly. The
highest and the lowest RBC mean values were reported among Los Angeles
and IVS-I-110 carriers respectively (ranges from 5.89 - 5.46 x 106
cells/mcL). The highest and the lowest Hb mean values were reported
among IVS-I-5 and -88 carriers respectively (ranges from 13.23 – 11.47
mg/dL). The highest and the lowest MCV mean values were reported among
codon 5 and IVS-II-1 carriers respectively (ranges from 69.12 – 64.59
fL). The highest and the lowest MCH mean values were reported among
IVS-I-5 and Los Angeles carriers respectively (ranges from 22.46 –
19.77 pg/cell). The highest and the lowest HbA2 mean values were
reported among IVS-II-1 and codon 5 carriers respectively (ranges from
4.72 – 3.80% of the total Hb). The highest and the lowest HbF mean
values were reported among IVS-I-5 and Los Angeles carriers
respectively (ranges from 3.16 – 0.26% of the total Hb). The highest
and the lowest HbA1 mean values were reported among IVS-I-110 and
IVS-I-5 carriers respectively (ranges from 94.92 – 83.26% of the total
Hb). Hence, these reported values while considering the SD, p-value, OR
and referring to the relevant guidelines can be helpful in offering a
hint to the local clinicians for more accurate referrals. This can also
be helpful as a boost for laboratory professionals for a more
straightforward ß-thalassemia testing guide in the region. We have also
found a significant difference regarding the MCV and the HbA2 levels
between the mutation types. Our findings mean that the type of mutation
causing ß-thalassemia has a high chance of affecting the MCV value and
HbA2 ratio. In addition, moving further into the details of mutations
impact on hematological indices we have illustrated that IVS-II-1 was
associated with increased risk for higher MCH and HbA2 in comparison to
other reported variants. It is also causing a lower MCV compared to
other mutations. Also, the codons 8/9 mutation was found to be
associated with significant increase in HbF values compared to other
mutations. That means that the IVS-II-1 variant has a high chance of
increasing the MCH and HbA2 while lowering the MCV when compared to
other mutation types. On the other hand, the presence of the codons 8/9
will probably raise the HbF proportion when compared to other
mutations. These findings act as a start point for more focused
interdisciplinary studies on the genomic and hematologic profile of
ß-thalassemia patients to find a more comprehensive map of
genotype-phenotype correlation.
Conclusions
Results
obtained from this study can help medical geneticists and other health
care professionals in the province to detect the carriers and their
at-risk fetuses through PND in a more rapid manner. Moreover, the idea
of existing a logical correlation between pathogenic HBB variants and
hematological indices can illuminate a new research topic of
investigation for similar future studies. Also, the idea of how the
mostly reported pathogenic variants might result in some
genotype-phenotype correlation in the region might assist the relevant
local clinicians for more rapid and accurate referrals to genetics
services.As previously discussed, ß-thalassemia is one of the most common genetic disorders worldwide and also in Iran.[3-14]
Although there are some treatments available for controlling and
recovering the disease such as routine blood transfusion (followed by
the iron chelation therapy), bone marrow transplantation and even gene
therapy, still genetic counseling and PND are known to be the best
available preventive options.[1,2] Authors hope the
current study will make a more accurate and useful guide for
ß-thalassemia diagnosis and prevention in the region.
Acknowledgment
This
study was performed on patients’ recourses in the ACECR medical
genetics laboratory of Mashhad as formerly addressed, based on medical
ethics and privacy policy regulations. Authors are thankful to the
medical genetics center of ACECR, the Mashhad branch, for their support
over this study.
References
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010 Dec;5(1):11. https://doi.org/10.1186/1750-1172-5-11 PMid: 20492708

- Campbell JS. Alpha and beta thalassemia. Am Fam Physician. 2009 Aug 15;80(4). PMid:19678601
- Irani
AD, Cheraghi Z, Bitaraf S, Cheraghi P, Safiri S. Prevalence of alpha
and beta-thalassemia mutations among carriers of thalassemia in
Shadegan city, southwest of Iran. Zahedan J Res Med Sci. 2015;17(8). https://doi.org/10.17795/zjrms1032
- Rahimi
Z, Raygani AV, Merat A, Haghshenass M, Gerard N, Nagel RL,
Krishnamoorthy R. Thalassemic mutations in southern Iran. Iran J Med
Sci. 2015 Aug 31;31(2).
- Rund
D, Filon D, Strauss N, Rachmilewitz EA, Oppenheim A. Mean corpuscular
volume of heterozygotes for beta-thalassemia correlates with the
severity of mutations. Blood. 1992 Jan 1;79(1):238-43. PMid:1728311
- Rezaee
AR, Banoei MM, Khalili E, Houshmand M. Beta-Thalassemia in Iran: new
insight into the role of genetic admixture and migration. Scientific
World Journal. 2012;2012. https://doi.org/10.1100/2012/635183 PMid: 23319887
- Mahdieh
N, Rabbani B. Beta thalassemia in 31,734 cases with HBB gene mutations:
pathogenic and structural analysis of the common mutations; Iran as the
crossroads of the Middle East. Blood Rev. 2016 Nov 1;30(6):493-508. https://doi.org/10.1016/j.blre.2016.07.001 PMid: 27453201
- Sarookhani
MR, Ahmadi MH, Amirizadeh N. Molecular Spectrum of Beta-Globin
Mutations in Transfusion-Dependent Patients with Thalassemia in Qazvin
Province, Iran. Iran J Med Sci. 2015 May 12;34(1):17-22.
- Sarookhani
MR, Ahmadi MH. Rare and unexpected beta thalassemic mutations in Qazvin
province of Iran. Afr J Biotechnol. 2010;9(1)
- Derakhshandeh-Peykar
P, Hourfar H, Heidari M, Kheirollahi M, Miryounesi M. The spectrum of
β-thalassemia mutations in Isfahan Province of Iran. Iran J Public
Health. 2008;37(2):106-11.
- Derakhshandeh-Peykar
P, Akhavan-Niaki H, Tamaddoni A, Ghawidel-Parsa S, Holakouie Naieni K,
Rahmani M, Babrzadeh F, Dilmaghani-Zadeh M, Daneshvar Farhud D.
Distribution of β-thalassemia mutations in the northern provinces of
Iran. Hemoglobin. 2007 Jan 1;31(3):351-6. https://doi.org/10.1080/03630260701462030
- Rahimi
F, Keikhani B, Aberumand M. Prenatal diagnosis (PND) of β-thalassemia
in the Khuzestan province, Iran. J Clin Diagn Res. 2007 Jan
1;1(6):454-9.
- Najmabadi
H, Ghamari A, Sahebjam F, Kariminejad R, Hadavi V, Khatibi T, Samavat
A, Mehdipour E, Modell B, Kariminejad MH. Fourteen-year experience of
prenatal diagnosis of thalassemia in Iran. Public Health Genomics.
2006;9(2):93-7. https://doi.org/10.1159/000091486 PMid: 16612059
- Abolghasemi
H, Amid A, Zeinali S, Radfar MH, Eshghi P, Rahiminejad MS, Ehsani MA,
Najmabadi H, Akbari MT, Afrasiabi A, Akhavan-Niaki H. Thalassemia in
Iran: epidemiology, prevention, and management. J Pediatr Hematol
Oncol. 2007 Apr 1;29(4):233-8. https://doi.org/10.1097/MPH.0b013e3180437e02 PMid: 17414565
- Langlois
S, Ford JC, Chitayat D, Désilets VA, Farrell SA, Geraghty M, Nelson T,
Nikkel SM, Shugar A, Skidmore D, Allen VM. Carrier screening for
thalassemia and hemoglobinopathies in Canada. J Obstet Gynaecol Can.
2008 Oct 1;30(10):950-9. https://doi.org/10.1016/S1701-2163(16)32975-9 PMid: 19038079
- Miller
SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting
DNA from human nucleated cells. Nucleic Acids Res. 1988 Feb
11;16(3):1215. https://doi.org/10.1093/nar/16.3.1215 PMid:3344216 PMCid:PMC334765
- Old
JM, Varawalla NY, Weatherall DJ. Rapid detection and prenatal diagnosis
of β-thalassaemia: studies in Indian and Cypriot populations in the UK.
Lancet. 1990 Oct 6;336(8719):834-7. https://doi.org/10.1016/0140-6736(90)92338-I PMid: 1976877
- Giardine
B, van Baal S, Kaimakis P, Riemer C, Miller W, Samara M, Kollia P,
Anagnou NP, Chui DH, Wajcman H, Hardison RC. HbVar database of human
hemoglobin variants and thalassemia mutations: 2007 update. Hum mutat.
2007 Feb 1;28(2):206. https://doi.org/10.1002/humu.9479 PMid: 17221864
 |
|
 |
|
 |
|
 |
|
[TOP]