Ferhat Arslan1, Sehnaz Alp2, Yahya Büyükasık3, Melda Comert Ozkan4, Fahri Şahin4, Seniha Basaran5, Arif Atahan Cagatay5, Ömer Haluk Eraksoy5, Kenan Aksu6, Barış Ertunç7, Volkan Korten8, Bahadır Ceylan9 and Ali Mert9.
1 Department
of Infectious Diseases and Clinical Microbiology, Faculty of Medicine,
Istanbul Medeniyet University, Istanbul, Turkey.
2 Department of Infectious Diseases and Clinical Microbiology, Faculty of Medicine, Hacettepe University, Ankara, Turkey.
3 Department of Hematology, Faculty of Medicine, Hacettepe University, Ankara, Turkey.
4 Department of Hematology, Faculty of Medicine, Ege University, İzmir, Turkey.
5 Department of Infectious Diseases and Clinical Microbiology, Faculty of Medicine, Istanbul University, Istanbul, Turkey.
6 Department of Internal Medicine, Division of Rheumatology, Faculty of Medicine, Ege University, İzmir, Turkey.
7
Department of Infectious Diseases and Clinical Microbiology, Faculty of
Medicine, Karadeniz Technical University, Trabzon, Turkey.
8 Department of Infectious Diseases and Clinical Microbiology, Faculty of Medicine, Marmara University, Istanbul, Turkey.
9
Department of Infectious Diseases and Clinical Microbiology, Faculty of
Medicine, Istanbul Medipol University, Istanbul, Turkey.
Correspondence to: Ferhat Arslan, MD. Department of Infectious Diseases
and Clinical Microbiology, İstanbul Medeniyet University Hospital,
Istanbul, Turkey. Tel.: +90 505 580 22 45. E-mail:
ferhatarslandr@hotmail.com
Published: September 1, 2018
Received: February 22, 2018
Accepted: July 11, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018047 DOI
10.4084/MJHID.2018.047
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Hemophagocytic
Lymphohistiocytosis (HLH) is an indicator of an exaggerated immune
response and eventually adverse outcomes. This study aimed to
investigate the clinical and laboratory features and outcomes of
patients with HLH. The medical records of 26 HLH adult patients (≥ 16
years of age) were retrospectively analyzed. Gender, age, the duration
of fever, time to diagnosis, etiology and laboratory data were
extracted from the records. The mean age was 38 ± 18 years, and 15
(58%) patients were female. A total of nine cases had infectious
diseases; four cases had rheumatologic diseases, three cases had
hematological malignancies while nine cases could not have a definitive
diagnosis. The median time to detection of HLH was 20 days (IQR: 8-30
d). Of the 25 patients, 11 (44%) died. The erythrocyte sedimentation
rates of the surviving and non-surviving patients were 39 ± 22 mm/h and
15 ± 13 mm/h, respectively. When a long-lasting fever is complicated by
bicytopenia or pancytopenia (especially), clinicians should promptly
consider the possibility of HLH syndrome to improve patients’
prognosis.
|
Introduction
Hemophagocytic
Lymphohistiocytosis (HLH) is a life-threatening clinical condition
caused by an exaggerated immune response that results in tissue
destruction.[1] Its primary form generally occurs due
to an underlying genetic immune dysfunction and primarily affects
infants and young children, whereas the secondary form occurs due to
various underlying conditions ranging from viral infections to
autoimmune diseases and cancers in adults.[2,3]
The
clinical features of HLH include fever, hepatosplenomegaly,
lymphadenopathy, neurological symptoms, and skin manifestations.
Cytopenia, elevated liver enzymes, high serum ferritin levels,
hypertriglyceridemia, and hypofibrinogenemia are laboratory
abnormalities that have frequently been reported in HLH patients.[4]
Although extremely elevated ferritin levels represent a highly
sensitive marker of HLH, this disease may be difficult to recognize and
diagnose.[5] Early diagnosis and prompt treatment of
the underlying cause are important to decrease morbidity and mortality
in patients with HLH.[6] In this study, we aimed to
investigate the clinical features and outcomes of patients with HLH in
tertiary care centers in Turkey through a retrospective chart review.
Materials and Methods
The
medical records of adult patients (≥ 16 years of age) diagnosed with
HLH between January 2010 and April 2016 at six university hospitals in
Turkey were reviewed retrospectively.
Infectious disease,
rheumatology, and hematology specialists were contacted for the
records. The Istanbul Medipol University Ethical Committee approved
this study.
All the patients who met the diagnostic guidelines of the Histiocyte Society (HLH-2004) were included in this study (Table 1).[7]
Molecular parameters, such as NK cell activity and soluble CD25, were
not available in this study due to a lack of laboratory facilities. The
following features were evaluated: fever (type, duration), time to
diagnosis, splenomegaly, hepatomegaly, lymphadenopathy, rash, serosal
involvement, respiratory symptoms, neurological symptoms, and
opportunistic infections. Hepatomegaly and splenomegaly were defined as
the long axis of the organs exceeding 155 mm and 130 mm on radiological
investigations, respectively.[8]
 |
Table 1. Hemophagocytic Lymphohistiocytosis 2004 Trial Diagnostic and Modified Criteria. |
Hemogram, ferritin,
lactate dehydrogenase (LDH), transaminase, bilirubin, triglyceride,
high-density lipoprotein (HDL), and fibrinogen levels and erythrocyte
sedimentation rates (ESRs) were analyzed. Hemophagocytosis was defined
as histological evidence of activated macrophages engulfing blood cells
in the bone marrow and/or other tissues.
Opportunistic infections
were defined as a new clinical condition with ongoing immunosuppression
due to HLH. When positron emission tomography with computed tomography
(PET/CT) was performed, the presence of fluorodeoxyglucose (FDG) uptake
was evaluated. Underlying triggering diseases and treatment modalities,
including types of initial therapy, secondary therapy, and adjunctive
therapy (supportive or underlying disease-specific treatment), were
evaluated. Patients without both ferritin and triglyceride levels in
their medical records were excluded.
The basic statistical
analysis was performed using R version 3.0.4. Results are expressed as
numbers (percentages) for categoric variable and as mean (standard
deviation) or median (interquartile range) for continuous variables.
The chi-square test and contingency table were used to compare
subgroups of patients. p values of less than 0.05 were considered as
statistically significant.
Results
26
patients met the inclusion criteria. The mean age of the patients was
38 years (range, 16-74), and 15 (58%) females were included. The
triggering etiologies were established in 17 cases (65%) and were as
follows: infection in 9 cases (Crimean-Congo hemorrhagic fever [CCHF]
in 1 case, Epstein-Barr virus [EBV] in 4 case, cytomegalovirus [CMV] in
1 case, influenza virus in 1 case, toxoplasmosis in 1 case, and
histoplasmosis in 1 case), rheumatologic disease in 3 cases
(adult-onset Still’s disease [AOSD] in 1 cases, rheumatoid arthritis in
1 case, and systemic lupus erythematosus [SLE] in 1 case), hematologic
malignancy in 3 cases (diffuse large B-cell lymphoma in 2 cases,
intravascular lymphoma in 1 case), and ulcerative colitis in 1 case. Of
the 26 patients, 11 (42%) died. Of the fatal cases, five had an
infectious etiology, one lymphoma, and the other five were associated
with an unknown etiology.
Fever was the most common sign among
the HLH patients. It was the primary presenting symptom in all cases.
The median duration of fever was 19 days (IQR, 10-30), and the median
time to diagnosis was 21 days (IQR 8-30). Hepatomegaly and splenomegaly
were detected in 21 (84%), and 23 (92%) patients, respectively, and 18
(72%) patients had lymphadenopathies (peripheral in 2 patients and
systemic in 16 patients).
Neurological manifestations, including
encephalopathy, seizures, and an altered level of consciousness, were
observed in nine cases (38%). Bilaterally thalamic involvement and
demyelinization findings were revealed in two different patients who
had not apparent neurological symptoms. In two patients, the
neurological findings were associated with herpes simplex virus (HSV)
reactivation. HSV DNA was detected in the cerebrospinal fluid (CSF) and
blood of two patients with encephalitis. Eight patients who received
immunosuppressive drugs for HLH developed concomitant opportunistic
infections (CMV in three patients, HSV in three patients, invasive
aspergillosis in one patient, and Pneumocystis jiroveci pneumonia in one patient).
The
erythrocyte sedimentation rates of the surviving and non-surviving
patients were 39 ± 22 mm/h and 16 ± 14 mm/h, respectively. The median
ferritin level was 8826 ng/ml (IQR 1656-27386 ng/ml, range 566-100000
ng/ml), the median LDH level was 1562 IU/ml (range; 342-5251 IU/ml) and
total bilirubin level median was 1.85 mg/dl (IQR 0.8-6.1 mg/dl). The
mean triglyceride level was 528±321 mg/dl, and the median HDL levels
were 7 mg/dl (IQR 5-12 mg/dl).
Bone marrow biopsy showed
hemophagocytosis in 22 of 26 (84%) patients. Additionally, there was
bone marrow involvement in two of the lymphoma cases. Activated
macrophages with maturation arrest (in 2 patients) and chronic
lymphoproliferation were the other findings in bone marrow biopsies.
Hemophagocytosis and hemosiderosis were observed in liver biopsies of
two of these patients.
Of the 26 patients, eight were evaluated by PET/CT scans; their findings are summarized in Table 2.
All
the patients with HLH received specific treatment for the
hemophagocytic syndrome. Three patients (12%) were treated only with
glucocorticoids, whereas the others received both glucocorticoids and
another drug(s) as initial treatment. The underlying diseases,
HLH-associated laboratory values and initial, secondary, and adjunctive
treatment modalities of the patients after HLH diagnosis are summarized
in Table 2.
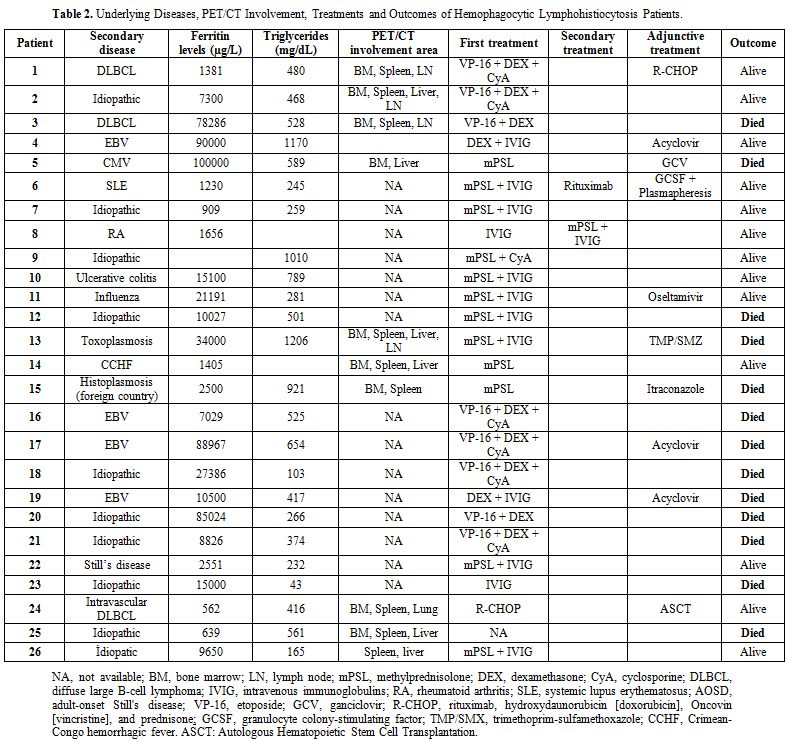 |
Table
2. Underlying Diseases, PET/CT Involvement, Treatments and Outcomes of Hemophagocytic Lymphohistiocytosis Patients. |
Discussion
Various underlying conditions that predispose patients to HLH have been defined in previous studies.[9,10,11] In children, underlying genetic defects play a predominant part in the development of HLH.[11,12] Although malignancies, frequently hematologic, seem to be the leading cause of adult-onset HLH,[11]
the predominant cause of HLH may differ by country because of different
genetic/ethnic backgrounds or differences in triggering agents,
particularly infections.[11,12] However, only 3 of 26
patients in our study were diagnosed with lymphoma. Although, this
finding may reflect the underrepresentation of hematology units at
participating centers or underdiagnosed lymphoproliferative diseases, a
similar distribution of causes of this disease in the Mediterranean
Region has been found in Spain but not in Italy and in France.[11]
Many
clinical and laboratory features were consistent with those reported
previously, e.g., high fever, cytopenia, splenomegaly, hepatomegaly,
and hyperferritinemia.[4] Fever was the primary presenting symptom in all of our patients, which is similar to a report from Riviere et al.[4]
Hyperferritinemia is a sensitive marker of HLH. No cutoff value has
been defined as critical; however, high levels of ferritin (> 50,000
µg/L) are associated with a poor prognosis.[13]
Hypofibrinogenemia is the main factor for blood cells velocity that
also stated as bad prognostic factor for HLH. Riviere et al. found
higher LDH levels in patients with hemophagocytic syndrome (positive
cases) than in negative and undetermined patients.[4]
In
our case series, the baseline ESR values, the median ferritin, LDH and
total bilirubin levels of the non-survivors were higher than those of
the survivors. But, due to limited numbers of patients to conclude
statistical significance with parametric and non-parametric tests, we
do not give any hypothetical test results here. We may say that more
tissue destruction like liver as cholestatic hepatitis and blood cells
hemolysis can explain the higher LDH and total bilirubin levels in
non-survivors as bad prognostic factors.
Neurological symptoms
may manifest as different clinical presentations, ranging from
depression and convulsions to progressive encephalopathy. In one study,
HSV reactivation was not found in patients with hemophagocytosis and
multi-organ failure.[14] In contrast, two of the five cases with neurological findings in our study involved HSV reactivation.
Hemophagocytosis
has been demonstrated in HLH patients, especially in the bone marrow,
spleen, liver, and lymph nodes. It is a diagnostic criterion.
Hemophagocytosis was reported in bone marrow aspirates in 84% of HLH
cases of the literature, which is similar to our findings.
Hemophagocytosis by itself is not a pathognomonic finding for the
diagnosis of HLH.[15] Because hemophagocytosis may be
a late finding in progressive HLH, repeat biopsies may be needed to
confirm the diagnosis in some cases.[1]
PET/CT is
a recently developed technique that is especially useful in cases of
malignancy, and this method may be valuable for diagnosing underlying
malignancies versus isolated HLH. In several reported cases, the
diffuse involvement of the bone marrow associated with spleen, liver,
and lymph node involvement has been found on PET/CT scans, which is
consistent with hemophagocytosis.[16] Little
information is available regarding whether PET/CT findings were related
to underlying or opportunistic diseases versus HLH. The underlying
conditions of our patients may have been responsible for the PET/CT
findings; however, one idiopathic case showed a diffuse involvement of
the reticuloendothelial system, which was considered to be associated
with HLH (Figure 1).
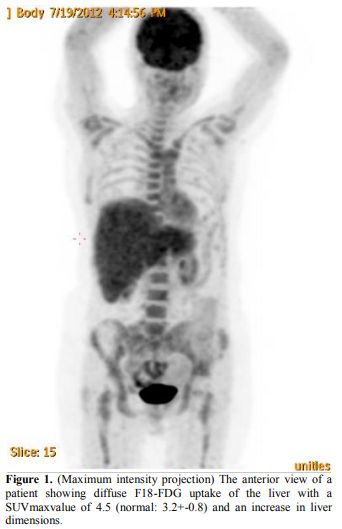 |
Figure 1. (Maximum
intensity projection) The anterior view of a patient showing diffuse
F18-FDG uptake of the liver with a SUVmaxvalue of 4.5 (normal:
3.2±0.8) and an increase in liver dimensions. |
In 2004, a new HLH treatment protocol was published (HLH-2004).[7]
Corticosteroids, cyclosporine, and etoposide constitute the backbone of
treatment. Among our cases, only two patients were treated with this
backbone therapy, and they recovered completely with good prognoses.
Etoposide-based steroid combination regimens have been confirmed to
improve HLH-associated outcomes in most trials.[17]
All
the patients had been treated with at least one antibiotic regimen
preceding the diagnosis of suspected sepsis (data not presented). Of
the 25 patients, 3 received only high-dose corticosteroids as initial
management, and only 1 case (a CCHF patient) resolved clinically. All
the patients with rheumatologic disease-related HLH responded to
treatment with good prognoses. While etoposide and cyclosporine
combination with high dose corticosteroid is the recommended regime for
HLH, in our study, eight patients were treated with etoposide
containing regimes, and six of them died. Mortality in rheumatological
diseases is significantly lower than that in infection- or
malignancy-related HLH.[18]
The reported
mortality of secondary HLH in adult case series varies from 20-74.8%,
which is similar to the rate of 44% in our case series.[4,11] Our cases with underlying rheumatological diseases also showed a good prognosis.
Advanced
age, the presence of lymphoma or infectious disease, transplantation,
and persistent fever within three days after the first treatment have
been clinically defined as poor prognostic factors.[4]
Although a statistical comparison was not possible, ten of the fatal
cases in this study had an infectious (~50%) and idiopathic (~50%)
etiology.
This retrospective case series study was intended to
present an additional adult HLH case series to the literature. Its
retrospective nature and small sample size are important limitations.
In conclusion, HLH occurs secondary to many diseases observed in
internal medicine practice. When a long-lasting fever is complicated by
bicytopenia or pancytopenia (especially), clinicians must promptly
consider the possibility of HLH syndrome since early diagnosis improves
patients’ prognosis. Especially low ESR levels and infectious or
idiopathic etiology may be alarmed conditions for unfavorable outcome
in the management of HLH.
References
- Rosado FGN, Kim AS: Hemophagocytic
lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin
Pathol 2013; 139:713–727 https://doi.org/10.1309/AJCP4ZDKJ4ICOUAT PMid:23690113

- Basheer
A, Padhi S, Boopathy V, Mallick S, Nair S, Varghese RG, Kanungo R.
Hemophagocytic Lymphohistiocytosis: an Unusual Complication of Orientia
tsutsugamushi Disease (Scrub Typhus). Mediterr J Hematol Infect Dis.
2015 ;7(1):e2015008. https://doi.org/10.4084/MJHID.2015.008. eCollection 2015.
- Hayden
A, Park S, Giustini D, et al.: Hemophagocytic syndromes (HPSs)
including hemophagocytic lymphohistiocytosis (HLH) in adults: A
systematic scoping review. Blood Rev 2016; 30:411–420 https://doi.org/10.1016/j.blre.2016.05.001 PMid:27238576
- Rivière
S, Galicier L, Coppo P, et al.: Reactive hemophagocytic syndrome in
adults: A multicenter retrospective analysis of 162 patients. Am J Med
2014 https://doi.org/10.1016/j.amjmed.2014.04.034 PMid:24835040
- Wormsbecker
AJ, Sweet DD, Mann SL, et al.: Conditions associated with extreme
hyperferritinemia (>3000 μg/L) in adults. Intern Med J 2015;
45:828–833 https://doi.org/10.1111/imj.12768 PMid:25851400
- Trottestam
H, Horne A, Aricò M, et al.: Chemoimmunotherapy for hemophagocytic
lymphohistiocytosis: long-term results of the HLH-94 treatment
protocol. Blood 2011; 118:4577–4584 https://doi.org/10.1182/blood-2011-06-356261 PMid:21900192 PMCid:PMC3208276
- Henter
J-I, Horne A, Aricó M, et al.: HLH-2004: Diagnostic and therapeutic
guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer
2007; 48:124–131 https://doi.org/10.1002/pbc.21039 PMid:16937360
- Dick R, Watkinson A: The liver and spleen. Textb Radiol Imaging 2002; 981–1028
- Song
Y, Pei R-J, Wang Y-N, et al.: Central Nervous System Involvement in
Hemophagocytic Lymphohistiocytosis in Adults: A Retrospective Analysis
of 96 Patients in a Single Center. Chin Med J (Engl) 2018; 131:776–783 https://doi.org/10.4103/0366-6999.228234 PMid:29578120 PMCid:PMC5887735
- Lorenz
F, Klimkowska M, Pawłowicz E, et al.: Clinical characteristics, therapy
response, and outcome of 51 adult patients with hematological
malignancy-associated hemophagocytic lymphohistiocytosis: a single
institution experience. Leuk Lymphoma 2018; 1–11 https://doi.org/10.1080/10428194.2017.1403018
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al.: Adult haemophagocytic syndrome. Lancet 2014; 383:1503–1516 https://doi.org/10.1016/S0140-6736(13)61048-X
- Ozen
S, Dai A, Coskun E, Oztuzcu S, Ergun S, Aktekin E, Yavuz S, Bay
A.Importance of hyperbilirubinemia in differentiation of primary and
secondary hemophagocytic lymphohistiocytosis in pediatric cases.
Mediterr J Hematol Infect Dis. 2014;6 :e2014067. doi:
10.4084/MJHID.2014.067. eCollection 2014 https://doi.org/10.4084/mjhid.2014.067
- Sackett
K, Cunderlik M, Sahni N, et al.: Extreme Hyperferritinemia: Causes and
Impact on Diagnostic Reasoning. Am J Clin Pathol 2016; 145:646–650 https://doi.org/10.1093/ajcp/aqw053 PMid:27247369
- François
B, Trimoreau F, Desachy A, et al.: [Hemophagocytosis during multiple
organ failure: M-CSF overproduction or viral reactivation?]. Ann Fr
Anesthèsie Rèanimation 2001; 20:514–519 https://doi.org/10.1016/S0750-7658(01)00410-5
- Ho
C, Yao X, Tian L, et al.: Marrow assessment for hemophagocytic
lymphohistiocytosis demonstrates poor correlation with disease
probability. Am J Clin Pathol 2014; 141:62–71 https://doi.org/10.1309/AJCPMD5TJEFOOVBW PMid:24343738
- Kim
J, Yoo SW, Kang S-R, et al.: Clinical implication of F-18 FDG PET/CT in
patients with secondary hemophagocytic lymphohistiocytosis. Ann Hematol
2014; 93:661–66. https://doi.org/10.1007/s00277-013-1906-y PMid:24061788
- Bergsten
E, Horne A, Aricó M, et al.: Confirmed efficacy of etoposide and
dexamethasone in HLH treatment: long-term results of the cooperative
HLH-2004 study. Blood 2017; 130:2728–2738 https://doi.org/10.1182/blood-2017-06-788349 PMid:28935695 PMCid:PMC5785801
- Ishii
E, Ohga S, Imashuku S, et al.: Nationwide survey of hemophagocytic
lymphohistiocytosis in Japan. Int J Hematol 2007; 86:58–65 https://doi.org/10.1532/IJH97.07012 PMid:17675268
[TOP]