Sreejesh Sreedharanunni1, Neelam Varma1, Man Updesh Singh Sachdeva1, Shano Naseem1, Pankaj Malhotra2, Deepak Bansal3, Amita Trehan3 and Subhash Varma2.
1 Department of Hematology, Postgraduate Institute of Medical Education and Research, Chandigarh, India - 160012.
2 Internal Medicine (Clinical Hematology), Postgraduate Institute of Medical Education and Research, Chandigarh, India -160012.
3 Pediatrics (Hematology/oncology unit), Postgraduate Institute of Medical Education and Research, Chandigarh, India -160012.
Correspondence to: Prof. Neelam Varma, MD. Professor and Head, Dept. of
Hematology, Fifth floor, Research Block A, Postgraduate Institute of
Medical Education and Research, Chandigarh, India -160012. Tel (O):
91-172-2755125 Fax: 91-172- 2747124. E-mail:
varmaneelam@yahoo.com
Published: September 1, 2018
Received: April 2, 2018
Accepted: July 20, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018052 DOI
10.4084/MJHID.2018.052
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Objective.
To determine the frequency, etiological spectrum and treatment outcome
of hypereosinophilia (HE) and hypereosinophilic syndromes (HES) in a
tropical setting.
Methods.
A retrospective analysis of hospital data of five years (January 2009
to December 2013) and a comprehensive prospective evaluation of
patients presenting with HE/HES over a period of 33 months (January
2014 to September 2016) was performed.
Results.
HE/HES was diagnosed in a total of 125 patients during the study period
with an estimated prevalence of 0.5-1 case per 100,000 population in
our hospital settings. 41 patients were excluded from the final
analysis due to lack of sufficient data. Infections, especially
helminths were the commonest cause (34%) followed by primary/clonal
HE/HES (24%) and reactive HE/HES secondary to various clonal disorders
(14.3%). A lymphocytic variant of HES and FIP1L1-PDGFRA positive HES
were diagnosed in 3.6% each. Imatinib-responsive BCR- ABL1 negative
HE/HES constitute 7.1% in our patients. None of the clinical or routine
laboratory features including the age of patients, duration of HE,
presence or absence of organomegaly, hemoglobin levels, eosinophil %,
absolute eosinophil count, total leukocyte count, platelet counts,
serum IgE levels or presence of myelofibrosis could predict or exclude
malignancy in patients with HE/HES. The absence of blasts in peripheral
blood or the absence of >5% blasts in bone marrow does not exclude
primary/clonal HES.
Conclusions.
An underlying malignancy (Primary HE/HES and neoplasms leading to
reactive HES; 35.7%) is diagnosed with nearly equal frequency compared
to infections (34.5%) in tropical settings. There are no hematological
or serological parameters, which can reliably be used to exclude an
underlying malignancy, necessitating a thorough follow-up and
comprehensive work-up in patients with HE/HES.
|
Introduction
Hypereosinophilia (HE) defined as >1.5 x 109/L
absolute eosinophil count (AEC) in peripheral blood and
hypereosinophilic syndrome (HES) defined as HE with organ dysfunction
are conditions associated with a wide spectrum of etiological factors
including infections, allergic and immunological disorders, drugs and
malignancies. Since its early descriptions,[1,2] there
were significant advances in the laboratory techniques resulting in the
identification of etiological factors in a large number of cases of
HES, otherwise categorized under idiopathic category. There has been
significant progress in the classification systems as well. While World
Health Organization (WHO) system[3] deals purely with
clonal causes, the definitions and classification proposed by
International Cooperative Working Group on Eosinophil Disorders
(ICOG-EO) appear to be a comprehensive system dealing with clonal and
non-clonal disorders.[4] The introduction of tyrosine
kinase inhibitors and other newer drugs in the treatment of HES is yet
another breakthrough in this field. Despite all progress, the diagnosis
and treatment of HES appear complicated due to various reasons. These
include a large number of secondary causes; the wide spectrum of
molecular abnormalities; co-occurrence of non-clonal and clonal causes
and the absence of any definite morphological or immunophenotypic
features differentiating clonal from non-clonal conditions. The
presence of numerous infective agents and the lack of availability of
laboratories performing a comprehensive workup of HES make it further
difficult in tropical countries. There is a scarcity of published data
on the HES from these regions. Knowledge of the spectrum of etiological
factors is an absolute necessity for generating a consensus opinion on
the essential investigations required, and for developing a management
protocol suitable for the socio-economic conditions prevalent in this
part of the world.
The aim of the study was to perform a
comprehensive clinico-pathological evaluation of cases of HE/HES over a
period of nearly eight years in an attempt to determine the relative
frequency and treatment outcome with a focus on
clonal/primary hypereosinophilia and secondary/reactive
hypereosinophilia associated clonal disorders.
Materials and Methods
The
study was conducted in the Department of Hematology in association with
the departments of adult clinical hematology and pediatric
hematology/oncology units from January 2009 to September 2016 (Total 93
months). The operational definition of hypereosinophilia used to
recruit patients in our study was >1.5 x 109/L
absolute eosinophil count (AEC) (and eosinophils >5%) in peripheral
blood. Eosinophil precursors were also included for calculating AEC.
The eosinophil % was determined by manually counting
a minimum of 200 leukocytes in the peripheral smear. The duration
of HE, presence or absence of clinical features related to organ
involvement was not considered for initial enrolment. The patients with
tissue HE in the absence of peripheral blood HE were excluded from the
study.
The retrospective analysis was performed using hospital
records of five years (January 2009 to December 2013). The clinical
features, laboratory findings, treatment and follow up details were
retrieved from the clinical record files for retrospective analysis. A
comprehensive prospective evaluation of patients presenting with HE/HES
was performed over a period of 33 months (January 2014 to September
2016).
Patients were evaluated with detailed history, clinical
examination, complete haemogram, serological tests (anti-nuclear
antibody/ANA, anti- neutrophil cytoplasmic antibody/ANCA, IgE levels,
parasite serology), skin hypersensitivity test for aspergillus, stool
examination, morphological evaluation of peripheral blood and bone
marrow, flow cytometry, reverse transcriptase PCR for TCF3-PBX1, ETV6-RUNX1, KMT2A-AFF1, RUNX1-RUNX1T1, CBFB-MYH11, BCR-ABL1, FIP1L1-PDGFRA, ETV6-PDGFRB translocations.[5–7] Amplification-refractory mutation system (ARMS) PCR for JAK2 V617F mutation[8] and fluorescent in situ hybridization (FISH) for PDGFRA, PDGFRB and FGFR1 gene rearrangements were also performed.[9]
The investigations were decided based on clinical findings, preliminary
investigations result as well as response to therapy. Serological tests
for parasites include IgM and IgG antibodies for Trichinella,
Toxoplasma, Toxocara, Echinococcus and antigen detection for
Microfilaria. These tests
detect both active and past infections, and the
interpretation depends on the titer and type of antibody positivity.
Patients were investigated in a stepwise manner to exclude reactive or
secondary causes of eosinophilia followed by evaluation for clonal
conditions.[10,11]
Six-color flow cytometry
(antibodies from BD Biosciences, BD Canto II flow cytometer and BD FACS
Diva software, San Jose, CA) was used for the evaluation of acute
leukemia and lymphoproliferative disorders. Peripheral blood was also
used to evaluate the presence of T cell subsets with abnormal
immunophenotype. T cell immunophenotype was simultaneously studied in
voluntary healthy control samples (n=25) with each batch of patients. A
minimum of 1 x 105 T cells was
acquired, gated for cytoplasmic CD3 and/or surface CD7 positive cells;
and analyzed for the presence of abnormal T cells. FISH was performed
using Vysis 4q12 tricolor rearrangement probe (Abbott molecular,
Illinois, USA), Vysis PDGFRB break - apart probe (Abbott molecular, Illinois, USA) and PoseidonTM Repeat FreeTM
FGFR1 (8p12) break-apart probe
(Kreatech biotechnology, Amsterdam, Netherlands)
respectively. A final categorization was attempted in each case
considering the clinical scenario, investigation results, follow up and
response to treatment.
The final categorization was based on
consensus classification by ICOG-EO. According to this classification,
HE (Peripheral blood absolute eosinophil count >1.5x109/L
without end- organ damage) is classified into hereditary/familial HE,
primary/clonal/neoplastic HE (eosinophils are clonal),
reactive/secondary HE (eosinophils are non-clonal or reactive) and HE
of undetermined significance. HES (HE as defined above with features of
end-organ damage attributable to HE) is classified into idiopathic HES
(no definite cause identified), primary/neoplastic HES and
secondary/reactive HES. In addition, the classification incorporates
two other categories (specific syndromes and several single-organ
restricted conditions associated with HE). Primary/neoplastic HE and
HES incorporates all the entities described in the WHO classification
of hematopoietic neoplasms.[4]
Discrete categorical data are presented as n
(%); continuous data given as median, range and interquartile range
(IQR). The comparison of two groups with skewed data was compared using
Mann Whitney test, and those with normally distributed data were
compared using student t- test. The comparison of categorical data
between two groups was performed by Chi-square test. All statistical
tests are two-sided and performed at a significance level of <0.05.
Statistical analysis was performed using SPSS for Windows (version
22.0; SPSS Inc., Chicago, IL, USA). All procedures followed were in
accordance with the ethical standards of the responsible committee on
human experimentation (institutional and national) and with the
Helsinki Declaration of 1975, as revised in 2008.
Results
A
diagnosis of HE/HES was made in a total of 125 patients over a period
of 93 months between January 2009 and September 2016. Among these, 41
patients were excluded from final analysis due to lack of
necessary work-up required for the final categorization. Excluded cases
include 11 patients of chronic myeloid leukemia (CML) on imatinib. Out
of the remaining 84 cases, 55 (65.5%) patients were enrolled
prospectively over a period of 33 months and in the remaining patients,
data were collected retrospectively. The demographic details and the
hematological findings of patients included in the final analysis are
summarized in Table 1.
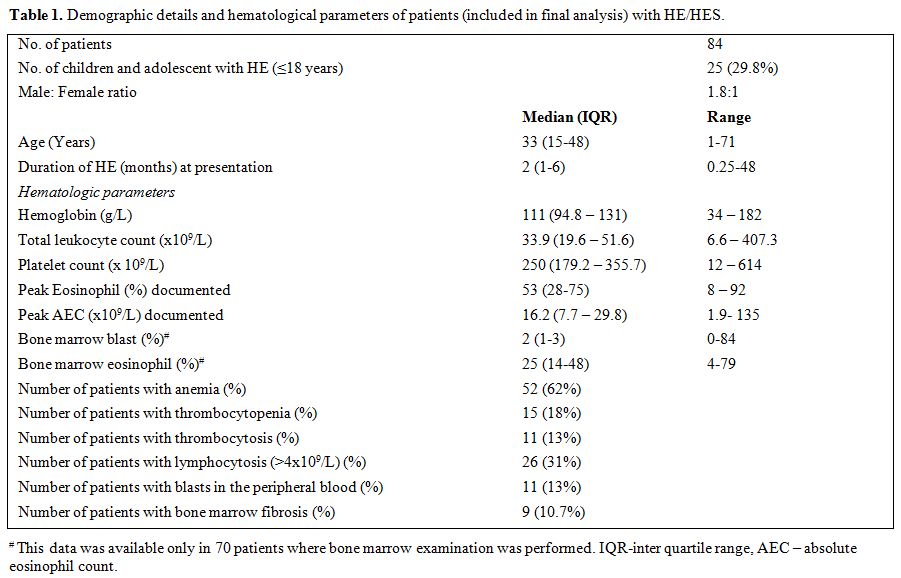 |
Table 1. Demographic details and hematological parameters of patients (included in final analysis) with HE/HES. |
The spectrum of HE/HES (n=84).
A final sub- categorization into HE, HES and specific syndromes
associated with HE as proposed by ICOG-EO classification system[4]
was attempted after considering the clinical data, laboratory findings,
and the treatment outcome. Majority of the patients (n=53; 63%) did not
have eosinophilia associated organ dysfunction (HE). Twenty-seven
patients (32%) were classified as HES, and another 5% had
specific syndromes/single organ disorder with HE [Eosinophilic
granulomatosis with polyangiitis/EGPA (n=2), Job syndrome (n=1),
Cutaneous eosinophilic vasculitis (n=1)]. Fourteen patients had
moderate eosinophilia defined as 1.5-5.0x109/L.[12]
Among these, ten patients (71.4%) had reactive HE/HES while rest of the
patients had clonal HE/HES. Rest of the patients (n=70; 83.3%) had
severe eosinophilia (>5x109/L). The etiologic spectrum associated with HE/HES is summarized in Figure 1 and Table 2.
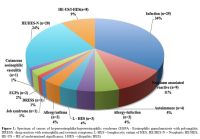 |
Figure 1. Spectrum of causes of
hypereosinophilia/hypereosinophilic syndrome (EGPA - Eosinophilic
granulomatosis with polyangiitis; DRESS - drug reaction with
eosinophilia and systemic symptoms; L-HES – lymphocytic variant of HES;
HE/HES-N – Neoplastic HE/HES; HE-US – HE of undetermined significance;
I-HES – idiopathic HES). |
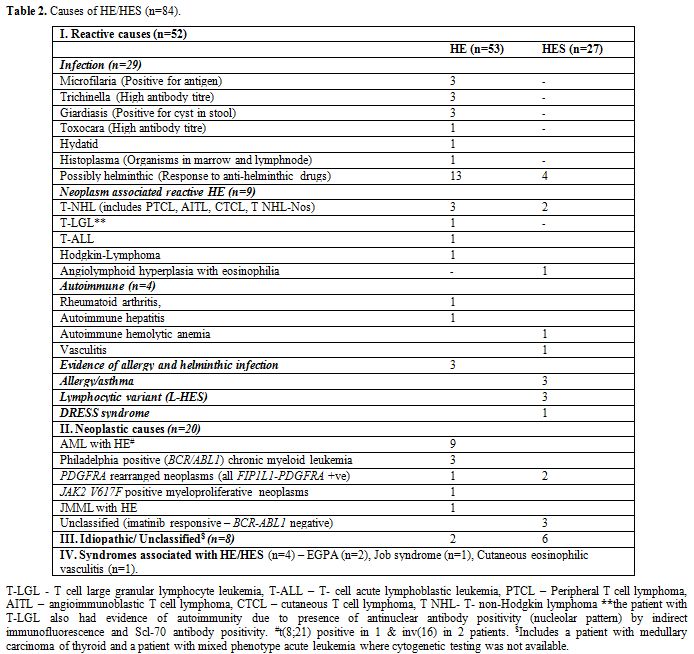 |
Table 2. Causes of HE/HES (n=84). |
Fever
was the most common symptom noted in 42% (n=35) of the patients. The
organ systems affected and the clinical features are
summarized in Table 3. Children and adolescents (≤ 18 years) constituted nearly 1/3rd
of the study population. In this group, infections (12/25; 48%) were
the commonest causes, followed by neoplasms (6/25; 24%). Autoimmune
(n=2) disorders, allergy (n=2), Job syndrome (n=1), drug reaction with
eosinophilia and systemic symptoms due to phenobarbital (DRESS) (n=1)
and EGPA (n=1) were other causes of HE/HES in this age group.
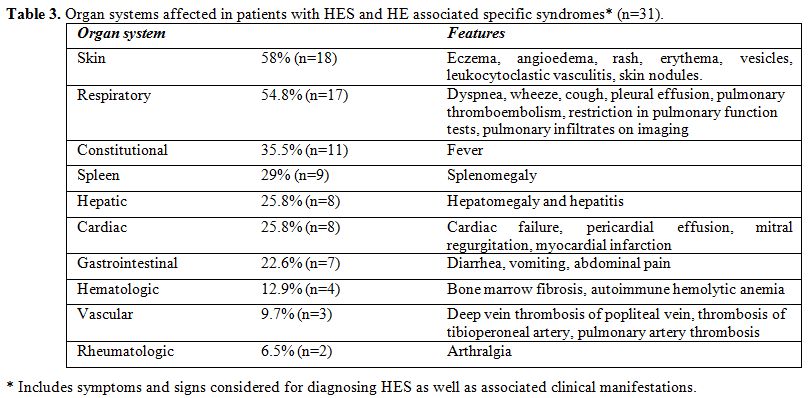 |
Table 3. Organ systems affected in patients with HES and HE associated specific syndromes* (n=31). |
Secondary HE/HES associated clonal disorders (n=12). HE was diagnosed as a reactive phenomenon secondary to various benign/malignant neoplasms in 10.7% (n=9) of patients (Table 2).
A lymphocytic variant of HE (L-HES) was diagnosed in another three
patients (3.6%), which constituted 11% of cases with HES. They were
diagnosed following demonstration of clones of mature T cells with
abnormal immunophenotype [CD3-CD4+CD5+ T cells (n=1) and CD4+CD8+T
cells (n=2)], a dramatic response of eosinophilia to steroids and the
requirement of long-term low dose steroids for controlling HE. However,
T cell receptor clonality studies were not performed.
Primary or Clonal or Neoplastic HE/HES (n=20).
A “clonal” HES (n=20; 23.8%) was diagnosed in the presence of either a
cytogenetic abnormality or bone marrow morphological evidence of a
myeloid neoplasm. Various causes are summarized in table 2. Overall imatinib responsive HE/HES (BCR-ABL1 negative) constitute 7.1% (n=6) of HE cases. Of these, FIP1L1-PDGFRA positive HE/HES was diagnosed in 3.6% (n=3) of patients (Figure 2).
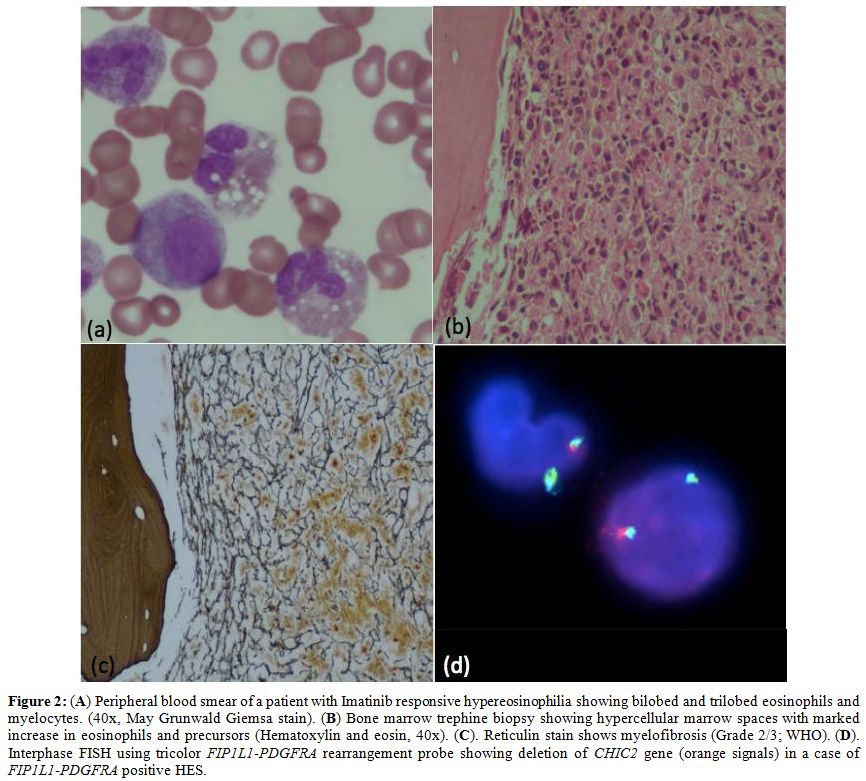 |
Figure 2. (A) Peripheral
blood smear of a patient with Imatinib responsive hypereosinophilia
showing bilobed and trilobed eosinophils and myelocytes. (40x, May
Grunwald Giemsa stain). (B) Bone marrow trephine biopsy showing
hypercellular marrow spaces with marked increase in eosinophils and
precursors (Hematoxylin and eosin, 40x). (C). Reticulin stain shows
myelofibrosis (Grade 2/3; WHO). (D). Interphase FISH using tricolor FIP1L1-PDGFRA rearrangement probe showing deletion of CHIC2 gene (orange signals) in a case of FIP1L1-PDGFRA positive HES. |
They were all males with age ranging from 25-40 years. The eosinophil % and AEC ranged from 54- 62% and 14.3-23 x 109/L
respectively. In one of them, HE was detected during the workup of
fever, while the other two patients presented with organ dysfunction
(cardiac failure and deep vein thrombosis). All of them had moderate
splenomegaly. Other three patients with HES were categorized as
imatinib responsive (BCR-ABL1
negative) HES as their symptoms (cardiac symptoms in two and bone
marrow fibrosis in the third patient) responded very well to low doses
(100mg) of this tyrosine kinase inhibitor. FIP1L1- PDGFRA was negative in one of these patients but was not tested in the other two cases due to its non-availability during that period.
HE with malignancy (n=30; 35.7%) vs. HE without malignancy (n=54; 64.3%).
HE was associated with malignancy as a reactive or clonal process in
35.7% of patients. Malignancy was diagnosed in 50% (7/14) of patients
with moderate eosinophilia, while it was diagnosed in 32.8% (23/70)
with severe eosinophilia. A comparison of various parameters was
performed between two groups of patients (HE with malignancy vs. HE
without malignancy; Table 4).
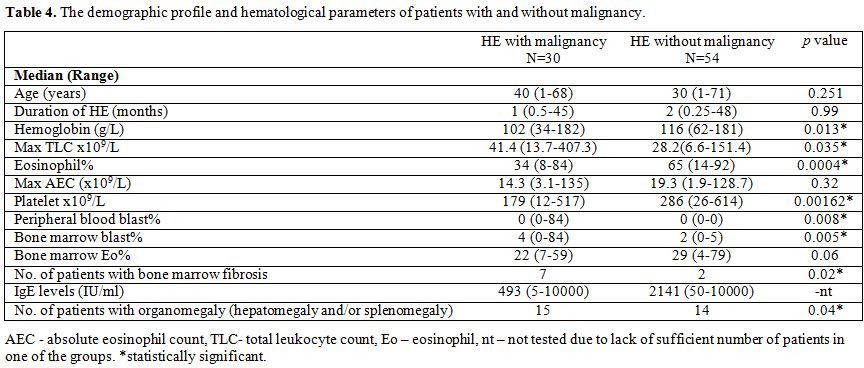 |
Table 4. The demographic profile and hematological parameters of patients with and without malignancy. |
Between
two groups, there was no significant difference for absolute eosinophil
count, age or duration of HE. HE with malignancy had significantly
lower hemoglobin levels (P=0.013), eosinophil % (P=0.0004) and platelet counts (P=0.0016); higher levels of total leukocyte count (P=0.035), higher frequency of bone marrow fibrosis (P=0.02) and organomegaly (P=0.04).
However, due to the significant overlap between groups, except for the
presence of blasts in peripheral blood and >5% blasts in the bone
marrow; none of the other clinical or routine laboratory features,
including the age of patients, duration of HE, presence or absence of
organomegaly, hemoglobin levels, eosinophil %, absolute eosinophil
count, total leukocyte count, platelet counts, serum IgE levels or
presence of myelofibrosis, could distinguish between patients with or
without malignancy.
Treatment and outcome of HE/HES. The treatment modalities employed are summarized in Figure 3.
Anti helminthic/parasitic medications including albendazole,
mebendazole, diethylcarbamazine (DEC), metronidazole and tinidazole
either alone or in combination; and steroids were the drugs most
commonly used. Imatinib was used in seven cases of which three patients
received it empirically with good response. Following treatment,
patients were followed up for the symptomatic response, response in AEC
and recurrence of HE. A follow up was available in 79.8% (67/84) of
patients, and the duration of follow-up ranged from 15 days to 58
months (median nine months). Among these, 94% (n=63) patients showed a
response to therapy with 13% (n=9) of them showing fluctuating
eosinophil levels. Two patients (AML with HE and idiopathic HE) expired
during the follow-up period, while another two patients (polycythemia
vera with HE and Job syndrome) continued to show HE during the last
follow-up. Twenty-four (35.8%) patients required long-term treatment
for the control of HE. These include patients with autoimmune disorders
[includes HE associated specific syndromes like EGPA) (n=5), L-HES
(n=3), idiopathic HES (n=3), allergy (n=3), CML (n=2), imatinib
responsive HE (n=3), FIP1L1-PDGFRA+ve
HES (n=2), cutaneous T cell lymphoma (n=1), T-cell large granular
lymphocyte leukemia (n=1) and unclassified HES (n=1)]. Oral or
inhalational low dose steroids (10 patients), other
immunosuppressants like azathioprine, cyclophosphamide, and
mycophenolate mofetil with/without steroids (n=7) and imatinib (n=7)
were given singly or in combination to these patients.
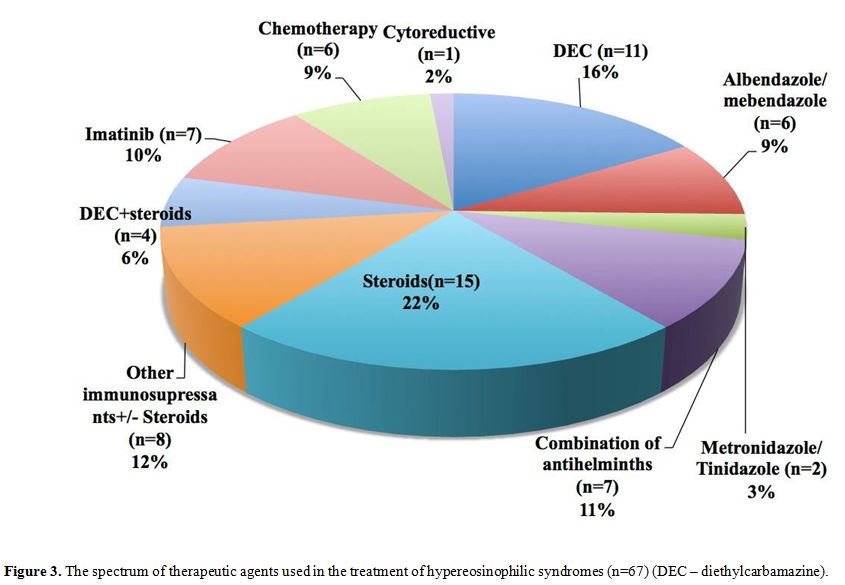 |
Figure 3. The spectrum of
therapeutic agents used in the treatment of hypereosinophilic syndromes
(n=67) (DEC – diethylcarbamazine). |
Discussion
The
estimation of prevalence of HE/HES is difficult due to the lack of
clear consensus and changes in its definition over the years. Based on
surveillance, epidemiology, and end result (SEER) database of the
National Cancer Institute, the age-adjusted incidence of HES is 0.18
per 100000.[13] These may not represent the true
incidence in tropical countries with high prevalence of parasitic
infections; and unfortunately, there is no data regarding the same from
several tropical countries including India. During our study period, we
encountered a total of 125 patients (over 93 months) with HE/HES. Since
the hospital received an average of 200,000 patients per month
(6000-7000 patients/day), this number translates to 0.5 to 1
case/100,000 hospital population. HES was diagnosed in 1/3rd
of these patients. The referral bias and the partially retrospective
nature of study make this figure likely to be an underestimate, and
HE/HES appears to be frequently encountered in this region.
The consensus proposal from Valent et al.[4]
has refined the definitions of HE and HES. However, the cut off levels
of HE and duration of disease is still arbitrary. Many patients with
AEC below the proposed levels and duration less than one month may
still require workup and treatment even in the absence of organ
dysfunction. This is especially important in regions with poor
socioeconomic conditions, where there is a difficulty in the follow-up
of patients. The present study includes all patients with HE/HES
irrespective of the duration. Overall, 63/84 (75%) of the patients had
persistent HE (≥ 1 month). In three patients with HES, duration of HE
was less than one month. Of these, one patient developed pulmonary
thromboembolism, myocardial infarction and expired within two weeks.
Other two patients presented with thrombosis and severe eczema
requiring intervention.
In our study, infections, especially
helminths were the commonest cause of HE as well as HES. We had
patients with malignancy (acute lymphoblastic leukemia, diffuse large B
cell lymphoma) and atopy/allergy, which was complicated by parasitic
infections. The diagnosis was predominantly based on the response to
anti- helminthic drugs (albendazole, mebendazole,
diethylcarbamazine/DEC) in others. However, a definite organism could
be demonstrated in only 12/29 (41.4%) patients with infection (Table 2).
It is challenging to demonstrate a definite organism in the majority of
the patients, which requires a wider panel and more sensitive
laboratory investigations. Moreover, many of these infections remain
subclinical with HE as the only manifestation. However, in symptomatic
patients, a detailed history of travel, exposure and the type of
symptom complexes will help to identify the cause of HE/HES.[14] In resource-limited settings, an empirical course of anti-helminthic therapy may safely precede detailed work-up.
HE
preceded the diagnosis of T lineage acute lymphoblastic leukemia,
Hodgkin Lymphoma and cutaneous T cell lymphoma in one patient each. HE
can mask the underlying neoplasm and may precede, occur simultaneously
or succeed in various neoplasms especially acute lymphoblastic
leukemia.[15-17] A thorough follow-up is must, and a
bone marrow examination and a detailed evaluation should not be delayed
in any patient with the slightest suspicion of an underlying neoplasm.
Three
patients (4%) showed HE associated with allergy/asthma. Another three
(4%) patients had evidence of both allergy and helminthic infection.
They showed a dramatic response to anti-helminthic drugs; however,
required low dose inhalational or oral steroids for control of
allergic/asthmatic symptoms. Allergic disorders are common in this part
of the world with the reported prevalence of approximately 30%. The
reported prevalence of asthma is 7.5%, and skin allergy is 5.8%. The
common precipitants of allergic disorders include dust, seasonal
changes, and food substances.[18] It is imperative to
take a detailed history of allergy and exposure to various allergens
while dealing with a case of HE/HES.
L-HES is a distinct variant of reactive HE[4,11]
characterized by the presence of HE in association with the secretion
of IL-5 from expanded immunophenotypically aberrant clonal T cells,
most commonly CD3-CD4+ T cells. The prevalence of L-HES in our study
was 3.6% (3/84) among all cases with HE or 11% (3/27) among cases with
HES. Since the first report of clonal proliferation of type 2 helper
cells in patient with HE,[19] there have been several
case reports and small series of cases describing this entity. The
reported prevalence ranges from 17-26%.[20-22]
Despite several reports and reviews, there is still a lack of consensus
in the diagnosis of this entity. Studies have used immunophenotype
abnormalities or presence of clonal T cell receptor rearrangements
alone[20,22] or both for diagnosing
L- HES. In our study, peripheral blood flow cytometry on normal
controls (n=25) and patients with various infections did not show
CD3-CD4+, CD4+CD8+ and CD2- T cells; but showed variable proportion of
CD4-CD8-, CD4+CD7-, CD7-, CD5- subsets of T cells as these may
represent NK cells, gamma delta T cells, NK/T cells or could be part of
the normal immunological response as seen in various infections.[23-25]
These normal alterations in the immunophenotype should be considered
before a diagnosis of L-HES is made especially in the absence of
clonality studies in tropical countries with high prevalence of
infections. Similarly, it is also important not to over diagnose L-HES
based on the isolated presence of clonal population of T cells without
immunophenotypic abnormalities as they can be identified in normal
population as well.[26]
Overlap HES refers
to patients with overlapping features of HES and EGPA or
those with single organ disease (like gastrointestinal disease,
episodic angioedema, eosinophilic fasciitis) with HE.[27]
We had three patients with overlap HES (two patients with EGPA and one
patient with cutaneous eosinophilic vasculitis). In these cases, the
cause-effect relationship, i.e. whether the HE causes single organ
dysfunction or, HE is a manifestation of the primary disease itself
remains debatable.
Among “clonal” HES, FIP1L1-PDGFRA+ve HES is the most common cause. Other PDGFRA, PDGFRB, FGFR1 and JAK2 related translocations are reported in very few (<5) patients or in single individuals.[28] In our study FIP1L1-PDGFRA+ HE/HES was diagnosed in 3.6% of patients compared to the reported frequency of 3-17% in various recent studies.[29-31] JAK2 V617F associated HE-N was seen in a single patient (1.2%) compared to the reported frequency of 4% in the literature.[30]
The presence of nearly 50 translocations and several mutations involving PDGFRA, PDGFRB, FGFR1, JAK2
and several other genes associated with clonal HES; and due to the
possibility of occurrence of various malignancies underlying reactive
HE/HES, it is imperative to identify markers which can predict
malignancy associated HES. This is especially important, as our study
shows that an underlying malignancy (n=30; 35.7%) in the form of clonal
HE/HES (n=20); Hodgkin or non-Hodgkin lymphomas and leukemias leading
to reactive HE/HES (n=9); or other miscellaneous malignancies like
carcinomas (n=1) causing paraneoplastic HE, is as frequent as various
infections (n=29; 34.5%). Our study shows that the patients with
malignancy had significantly lower Hb levels, eosinophil %, and
platelet counts; higher levels of total leukocyte count (TLC),
peripheral blood or bone marrow blast %, the incidence of bone marrow
fibrosis and a higher proportion of patients with organomegaly. The
median IgE levels were also higher in patients without malignancy.
However, the overlap in the values between the two groups makes them
less useful to predict or exclude malignancy. Again, though the
presence of blasts in peripheral blood and >5% blasts in the bone
marrow was exclusively seen in malignancy; their absence did not
exclude clonal HE/HES. Blasts, mast cells, and fibrosis are reported to
be more frequent in bone marrow biopsies of patients with FIP1L1- PDGFRA translocation.[32] But, none of our patients with FIP1L1-PDGFRA
translocation had blasts in the peripheral blood or bone marrow blasts
>5%. Only one of them had anemia while platelet counts were normal
in all of them. However, 50% of Imatinib responsive HES (BCR-ABL1
negative) patients had myelofibrosis, probably the most useful
morphological indicator in our study. Elevated serum vitamin B12 and
tryptase levels are other parameters suggested to be associated with
myeloproliferative neoplasms[33] but were not evaluated in the current study.
An
exact categorization could not be possible in eight patients (HE of
undetermined significance/Idiopathic HES/unclassified) due to various
reasons. The inclusion of cytogenetic
testing, a complete panel of FISH testing for
PDGFRA, PDGFRB, FGFR1, JAK2
rearrangements and molecular testing for clonal T cell clones might
reveal a cause in many of these cases. The improvement in the knowledge
about pathobiology of HE/HES and availability of advanced laboratory
technology including next-generation sequencing is expected to solve
the mystery behind several cases of idiopathic HES/HE of undetermined
significance in future. Another limitation of our study is the
compilation of retrospective with prospective data probably resulting
in selection bias and underestimating the true prevalence of HE/HES.
Compared to two large studies from National Institute of Health[27] and Mayo Clinic[34]
respectively, our study shows very high frequency of secondary/reactive
HE/HES (10% and 46% vs. 62%) and neoplastic/clonal/myeloproliferative
HE/HES (10% and 17% vs. 24%), very low frequency of idiopathic HE/HES
(47% and 32% vs. 9.8%) and low prevalence of L-HES (14.8% and 4% vs.
3.6%).
Conclusion
HE/HES
appears to be an under-reported public health problem in tropical
settings with an estimated prevalence of 0.5-1- case/100,000 population
in hospital settings. Infections especially helminths are the commonest
cause of HE/HES in our study, and should be excluded even in patients
with other causes of HE. The spectrum of infections is so wide that the
demonstration of the specific infective agent is often difficult in
resource-limited settings; necessitating an empirical course of
anti-helminths in most of the patients. In contrary to the general
perception in tropical countries, an underlying malignancy is diagnosed
with nearly equal frequency compared to infections. An underlying
malignancy is highly likely in patients with presence of blasts in
peripheral blood, >5% blasts in bone marrow and bone marrow
fibrosis. But there are no hematological or serological parameters,
which can reliably be used to exclude an underlying malignancy,
necessitating a thorough follow-up and comprehensive work-up in
patients with HE/HES.
Acknowledgments
The authors are thankful to Mrs. Praveen Bose for the technical help in performing immunophenotypic studies.
References
- Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1977;54:1–27. https://doi.org/10.1097/00005792-197501000-00001

- Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann Intern Med 968;68:1220–9. https://doi.org/10.7326/0003-4819-68-6- 1220
- Swerdlow
SH CE, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J,
Arber DA, Hasserjian RP, Le Beau MM, Orazi A, Seibert R (editors). WHO
Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC:
Lyon; 2017
- Valent
P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, Hellmann A,
Metzgeroth G, Leiferman KM, Arock M, Butterfield JH, Sperr WR, Sotlar
K, Vandenberghe P, Haferlach T, Simon HU, Reiter A, Gleich GJ.
Contemporary consensus proposal on criteria and classification of
eosinophilic disorders and related syndromes. J Allergy Clin Immunol.
2012;130:607–612.e9 https://doi.org/10.1016/j.jaci.2012.02.019 PMid:22460074 PMCid:PMC4091810
- Bhatia
P, Binota J, Varma N, Marwaha R, Malhotra P, Varma S. Incidence of
Common Fusion Transcripts in Adult and Pediatric Acute Myeloid Leukemia
(AML) Cases: Experience of a Tertiary Care Research Institute. Mediterr
J Hematol Infect Dis. 2012;4:e2012042. https://doi.org/10.4084/mjhid.2012.042 PMid:22811791 PMCid:PMC3395706
- Bhatia
P, Binota J, Varma N, Bansal D, Trehan A, Marwaha RK, Malhotra P, Varma
S. A Study on the Expression of BCR-ABL Transcript in Mixed Phenotype
Acute Leukemia (MPAL) Cases Using the Reverse Transcriptase Polymerase
Reaction Assay (RT-PCR) and its Correlation with Hematological
Remission Status Post Initial Induction Therapy. Mediterr J Hematol
Infect Dis. 2012;4:e2012024. https://doi.org/10.4084/mjhid.2012.024 PMid:22708039 PMCid:PMC3375663
- Bhatia
P, Binota J, Varma N, Bansal D, Trehan A, Marwaha RK, Malhotra P, Varma
S. Incidence of common chimeric fusion transcripts in B-cell acute
lymphoblastic leukemia: an Indian perspective. Acta Haematol
2012;28:17–9. https://doi.org/10.1159/000338260 PMid:22572394
- Kui
JS, Espinal-Witter R, Wang YL. Laboratory detection of JAK2V617F in
human myeloproliferative neoplasms. Methods Mol Biol. 2013;999:41–57. https://doi.org/10.1007/978-1-62703-357-2_3 PMid:23666689
- Knoll
JM, Lichter P. In situ hybridization to metaphase chromosomes and
interphase nuclei. In: Haines JL, editor. Current Protocols in Human
Genetics. John Wiley & Sons, Inc, Hoboken, NJ, 2005;p.
4.3.1-4.3.31. https://doi.org/10.1002/0471142905.hg0403s45
- Gotlib
J. World Health Organization-defined eosinophilic disorders: 2014
update on diagnosis, risk stratification, and management. Am J Hematol.
2014;89:325–37. https://doi.org/10.1002/ajh.23664 PMid:24577808
- Gotlib
J. World Health Organization-defined eosinophilic disorders: 2017
update on diagnosis, risk stratification, and management. Am J Hematol.
2017;92:1243–59. https://doi.org/10.1002/ajh.24880 PMid:29044676
- Fulkerson PC, Rothenberg ME. Targeting Eosinophils in Allergy, Inflammation and Beyond. Nat Rev Drug Discov. 2013;12:117-29 https://doi.org/10.1038/nrd3838 PMid:23334207 PMCid:PMC3822762
- Crane
MM, Chang CM, Kobayashi MG, Weller PF. Incidence of myeloproliferative
hypereosinophilic syndrome in the United States and an estimate of all
hypereosinophilic syndrome incidence. J Allergy Clin Immunol.
2010;126:179–81. https://doi.org/10.1016/j.jaci.2010.03.035 PMid:20639012 PMCid:PMC5781228
- O'Connell EM, Nutman TB. Eosinophilia in Infectious Diseases. Immunol Allergy Clin North Am. 2015;35:493–522. https://doi.org/10.1016/j.iac.2015.05.003 PMid:26209897 PMCid:PMC4515572
- Song
G, Liu H, Sun F, Gu L, Wang S. Acute lymphocytic leukemia with
eosinophilia: a case report and review of the literature. Aging Clin
Exp Res. 2012;24:555–8. PMid:22510980
- Kaneko
H, Shimura K, Yoshida M, Ohkawara Y, Ohshiro M, Tsutsumi Y, Iwai T,
Horiike S, Yokota S, Taniwaki M. Acute lymphoblastic leukemia with
eosinophilia lacking peripheral blood leukemic cell: a rare entity.
Indian J Hematol Blood Transfus. 2014;30:80–3. https://doi.org/10.1007/s12288-013-0255-2 PMid:25332543 PMCid:PMC4192183
- Chien
AJ, Argenyi ZB, Colven RM, Kirby P. Acute lymphoblastic leukemia
presenting with urticarial plaques and hypereosinophilia in a child. J
Am Acad Dermatol. 2004;51:S151-155. https://doi.org/10.1016/j.jaad.2004.04.018 PMid:15577757
- Nitin
J, Palagani R, Shradha N, Vaibhav J, Kowshik K, Manoharan R, Nelliyanil
M. Prevalence, severity and risk factors of allergic disorders among
people in south India. Afr Health Sci. 2016;16:201– 9. https://doi.org/10.4314/ahs.v16i1.27 PMid:27358633 PMCid:PMC4915438
- Cogan
E, Schandené L, Crusiaux A, Cochaux P, Velu T, Goldman M. Brief report:
clonal proliferation of type 2 helper T cells in a man with the
hypereosinophilic syndrome. N Engl J Med. 1994;330:535–8. https://doi.org/10.1056/NEJM199402243300804 PMid:8302319
- Ogbogu
PU, Bochner BS, Butterfield JH, Gleich GJ, Huss-Marp J, Kahn JE,
Leiferman KM, Nutman TB, Pfab F, Ring J, Rothenberg ME, Roufosse F,
Sajous MH, Sheikh J, Simon D, Simon HU, Stein ML, Wardlaw A, Weller PF,
Klion AD. Hypereosinophilic syndrome: a multicenter, retrospective
analysis of clinical characteristics and response to therapy. J Allergy
Clin Immunol. 2009;124:1319– 1325.e3. https://doi.org/10.1016/j.jaci.2009.09.022 PMid:19910029 PMCid:PMC2829669
- Roufosse F. Hypereosinophilic syndrome variants: diagnostic and therapeutic considerations. Haematologica. 2009;94:1188–93. https://doi.org/10.3324/haematol.2009.010421 PMid:19734412 PMCid:PMC2738708
- Helbig
G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien
S. T-cell abnormalities are present at high frequencies in patients
with hypereosinophilic syndrome. Haematologica. 2009;94:1236–41 https://doi.org/10.3324/haematol.2008.005447 PMid:19734416 PMCid:PMC2738715
- Roden
AC, Morice WG, Hanson CA. Immunophenotypic attributes of benign
peripheral blood gammadelta T cells and conditions associated with
their increase. Arch Pathol Lab Med. 2008;132:1774–80. PMid:18976014
- Morice
WG. The immunophenotypic attributes of NK cells and NK- cell lineage
lymphoproliferative disorders. Am J Clin Pathol. 2007;127:881–6. https://doi.org/10.1309/Q49CRJ030L22MHLF PMid:17509985
- Posnett
DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal
elderly humans: the T cell equivalent to "benign monoclonal
gammapathy." J Exp Med. 1994;79:609–18. https://doi.org/10.1084/jem.179.2.609
- Reinhold U, Abken H. CD4+ CD7- T cells: a separate subpopulation of memory T cells? J Clin Immunol. 1997;17:265–71. https://doi.org/10.1023/A:1027318530127 PMid:9258765
- Williams
KW, Ware J, Abiodun A, Holland-Thomas NC, Khoury P, Klion AD.
Hypereosinophilia in Children and Adults: A Retrospective Comparison. J
Allergy Clin Immunol Pract. 2016;4:941–947.e1. https://doi.org/10.1016/j.jaip.2016.03.020 PMid:27130711 PMCid:PMC5010485
- Valent
P, Horny H-P, Bochner BS, Haferlach T, Reiter A. Controversies and open
questions in the definitions and classification of the
hypereosinophilic syndromes and eosinophilic leukemias. Semin Hematol.
2012;49:171–81. https://doi.org/10.1053/j.seminhematol.2012.01.009 PMid:22449627
- Pardanani
A, Brockman SR, Paternoster SF, Flynn HC, Ketterling RP, Lasho TL, Ho
CL, Li CY, Dewald GW, Tefferi A. FIP1L1-PDGFRA fusion: prevalence and
clinicopathologic correlates in 89 consecutive patients with moderate
to severe eosinophilia. Blood. 2004;104:3038– 45. https://doi.org/10.1182/blood-2004-03-0787 PMid:15284118
- Schwaab
J, Umbach R, Metzgeroth G, Naumann N, Jawhar M, Sotlar K, Horny HP,
Gaiser T, Hofmann WK, Schnittger S, Cross NC, Fabarius A, Reiter A. KIT
D816V and JAK2 V617F mutations are seen recurrently in
hypereosinophilia of unknown significance. Am J Hematol. 2015;90:774–7.
https://doi.org/10.1002/ajh.24075 PMid:26017288
- Roche-Lestienne
C, Lepers S, Soenen-Cornu V, Kahn JE, Laï JL, Hachulla E, Drupt F,
Demarty AL, Roumier AS, Gardembas M, Dib M, Philippe N, Cambier N,
Barete S, Libersa C, Bletry O, Hatron PY, Quesnel B, Rose C, Maloum K,
Blanchet O, Fenaux P, Prin L, Preudhomme C. Molecular characterization
of the idiopathic hypereosinophilic syndrome (HES) in 35 French
patients with normal conventional cytogenetics. Leukemia.
2005;19:792–8. https://doi.org/10.1038/sj.leu.2403722 PMid:15772698
- Schwaab
J, Jawhar M, Naumann N, Schmitt-Graeff A, Fabarius A, Horny HP, Cross
NC, Hofmann WK, Reiter A, Metzgeroth G. Diagnostic challenges in the
work up of hypereosinophilia: pitfalls in bone marrow core biopsy
interpretation. Ann Hematol. 2016;95:557–62. https://doi.org/10.1007/s00277-016-2598-x PMid:26797429
- Klion AD. How I treat hypereosinophilic syndromes. Blood. 2015;126:1069–77. https://doi.org/10.1182/blood-2014-11-551614 PMid:25964669 PMCid:PMC4551360
- Lim
K-H, Tefferi A, Li CY, Pardanani AD. Hypereosinophilia in 357
Consecutive Patients: Disease Spectrum and Clinical and Laboratory
Correlates. Blood. 2009;114:3903–3903.
[TOP]
