Attawut Chaibunruang1, Kanda Sornkayasit1, Mattanee Chewasateanchai2, Peerayoot Sanugul2, Goonnapa Fucharoen1 and Supan Fucharoen1.
1 Centre for
Research and Development of Medical Diagnostic Laboratories, Faculty of
Associated Medical Sciences, Khon Kaen University, Khon Kaen, Thailand.
2 Regional Health Promotion Center 7, Khon Kaen, Thailand.
Correspondence to: Goonnapa Fucharoen or Supan Fucharoen. Centre for
Research and Development of Medical Diagnostic Laboratories, Faculty of
Associated Medical Sciences, Khon Kaen University, Khon Kaen, Thailand
40002, Tel/Fax +66-43-202083. E-mail:
goonnapa@kku.ac.th or
supan@kku.ac.th
Published: September 1, 2018
Received: April 27, 2018
Accepted: July 28, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018054 DOI
10.4084/MJHID.2018.054
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background.
To provide accurate prevalence information of thalassemia in northeast
Thailand, authors performed thalassemia screening in newborns after 20
years implementation of a prevention and control program.
Methods.
Study was done on 350 cord blood specimens collected consecutively at
Maternal and Child Hospital, Regional Health Promotion Center 7, Khon
Kaen, Thailand. All kinds of α- and β-thalassemias were identified using combined hemoglobin (Hb) and DNA analyses.
Results.
Among 350 newborns examined, subjects with thalassemia genes were
identified in 184 (52.6%) cases with as many as 22 different genotypes.
The most prevalent one was Hb E (39.1%). The incidence of 3.1% α0-thalassemia, 25.9% α+-thalassemia,
5.4% Hb Constant Spring and 1.4% of Hb Paksé were encountered.
Heterozygous β-thalassemia was found in 2 cases (0.6%). Hb capillary
electrophoresis could demonstrate Hb E in all cases with Hb E and
detected different levels of Hb Bart’s for different α-thalassemia genotypes but not in all cases with α-thalassemia. No newborn with severe thalassemia diseases was encountered.
Conclusion. This study reveals that α-thalassemia,
β-thalassemia, and Hb E carriers as well as complex thalassemia
syndromes are still prevalence and indicates a need for continuing a
prevention and control program in the region.
|
Introduction
Thalassemia
and hemoglobinopathies are very heterogeneous and are major public
health problems in Southeast Asian countries. In Thailand, the
prevalence based statistically on phenotype has been estimated at
2.5-10% for α0-thalassemia (--SEA and --THAI), 1-8% for hemoglobin (Hb) Constant Spring and Hb Paksé and 15-20% for α+-thalassemia (-α3.7 and -α4.2) and 3-9% for β-thalassemia. Hb E can be found between 30-50% especially in the northeastern part of the country.[1,2]
It is estimated that around 1% of the Thai population has thalassemia
disease and each year there are more than 12,000 new births with
thalassemia syndromes. With the high prevalence and diverse
heterogeneity of thalassemia and hemoglobinopathies, around 60
thalassemia syndromes are encountered in Thailand.[3]
Accordingly, the national prevention and control program has been
established throughout the country under the support of the National
Health Security Office (NHSO). Under this program, all pregnant women
and their husbands can have thalassemia screening and diagnostics at
the government hospitals for free. However, with the reasons on
clinical severity and budget for treatment of each thalassemia syndrome
and limited laboratory facilities and economic resources, the program
has been focused mainly on the three severe thalassemia diseases
including homozygous α0-thalassemia (Hb Bart’s hydrops fetalis), homozygous β-thalassemia and Hb E-β-thalassemia.[4]
The main objective of the program is to prevent births of new cases
with these three severe thalassemia diseases. Carrier screening,
genetic counseling, and prenatal diagnosis are offered to couples at
risk of having fetuses with these three severe thalassemia syndromes.[5,6]
At our center in northeast Thailand, this prevention and control
program has been implemented since 1993. A retrospective analysis to
evaluate the overall performance of the prevention and control service
at the center during 1993-2008 has demonstrated a satisfactory
prevention outcome.[7] To provide accurate data on the
current prevalence of thalassemia after 20 years of a prevention and
control program, we have now looked prospectively on 350 newborns and
determined thalassemia genotypes using complete Hb and molecular
investigations.
Materials and Methods
Subjects.
The study protocol was approved by our institutional review board (IRB)
of Khon Kaen University, Khon Kaen, Thailand (HE 542253). Informed
consent was obtained from the parents. Based on the prevalence of
thalassemia found in the region,[8] the sample size was estimated at 334.[9].
Therefore, cord blood specimens (n=350) anti-coagulated with EDTA were
consecutively collected from babies delivered at the Maternal and Child
Hospital, Regional Health Promotion Center 7, Khon Kaen province,
Northeast Thailand during January to May 2012. Before collection, the
umbilical cord was wiped with gauze to reduce maternal blood
contamination. Preterm newborns and newborns with other abnormalities
were excluded.
Hb analysis and DNA analysis.
Hb fractions and quantifications were performed using automated
capillary electrophoresis (CE) (Capillarys 2 Flex Piercing: Sebia,
Lisses, France), applying the manufacturer protocol.[10] Genomic DNA was prepared from leukocytes using the standard method. Identification of the α0-thalassemia (--SEA and --THAI) and α+-thalassemia
(3.7 and 4.2 kb deletions) were routinely performed in our laboratory
using gap-PCR. Hb Constant Spring and Hb Paksé were identified using
multiplex allele-specific PCR.[2,7,11,12,13] Screening for α-globin gene triplication (αααanti3.7) was done using a PCR method as previously described.[14] Common β-thalassemia genes found in Thailand were examined using allele-specific PCR assays.[15]
Results
Among
the 350 babies examined, no thalassemia was detected in 166 cases
(47.4%). Based on Hb and DNA analyses, the remaining 184 cases (52.6%)
were found to carry thalassemia genes with as many as 22 thalassemia
genotypes including those with double or triple heterozygosities as
shown in Table 1. However, no case with severe thalassemia diseases targeted in a prevention and control program including homozygous α0-thalassemia,
homozygous β-thalassemia, and Hb E-β-thalassemia was encountered. As
expected, the three most common prevalent thalassemias were
heterozygous Hb E which was found in 60 cases (17.1%) followed by
heterozygous α+-thalassemia (-α3.7) (11.4%) and double heterozygous α+-thalassemia (-α3.7) with Hb E (5.7%). Two cases of heterozygous β0-thalassemia
(codon 17; A-T) were identified. Other genotypes were observed at lower
frequencies. The corresponding number of allele detected, gene
frequency and incidence of each thalassemic gene is shown in comparison
with those described in other areas of Thailand in Table 2.[16-18]
As shown the table, the overall incidences of 35.8% for all forms of
α-thalassemia, 39.1% Hb E and 0.6% β-thalassemia were encountered in
newborns in this study
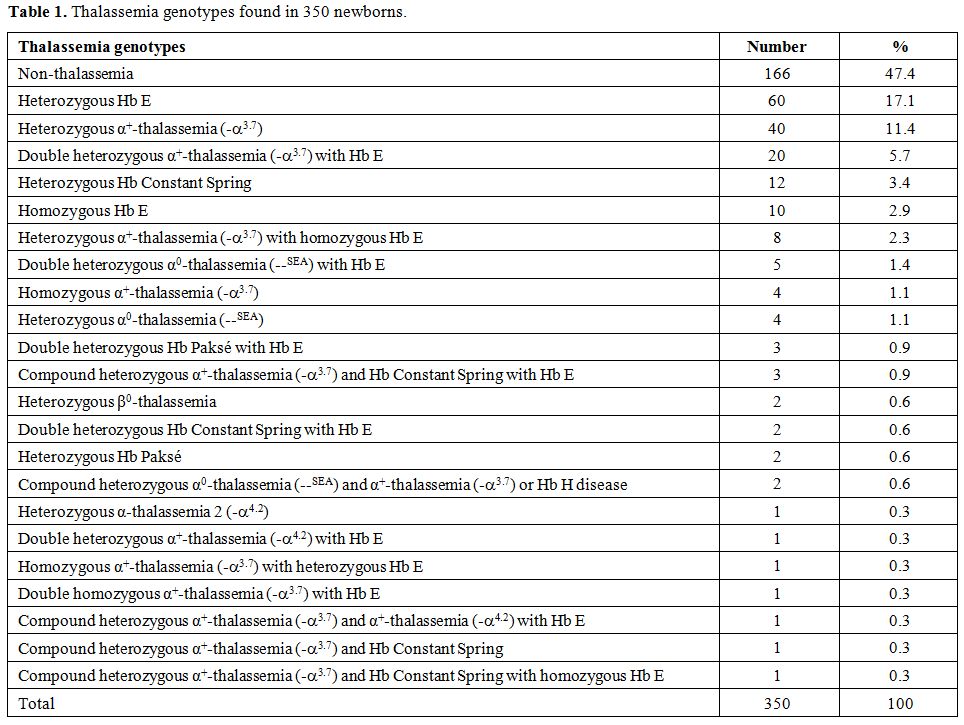 |
Table 1.
Thalassemia genotypes found in 350 newborns. |
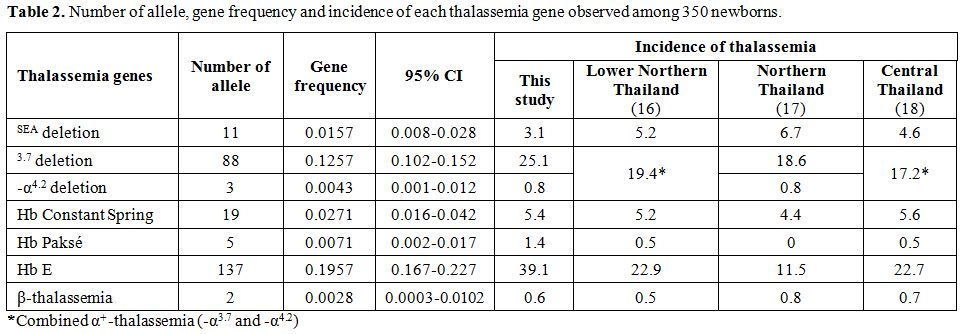 |
Table 2. Number of allele, gene frequency and incidence of each thalassemia gene observed among 350 newborns. |
Twelve Hb patterns and corresponding genotypes of the newborns are shown in Table 3. A normal Hb FA pattern was identified in 183 cases including 145 normal babies, 33 α+-thalassemia carriers (-α/αα), four carriers of non-deletion α+-thalassemia (αTα/αα) and a baby with β-thalassemia heterozygote (αα/αα, β17/βA).
Hb Bart’s was detected in most of the babies with α-thalassemia, i.e.,
46 of 48 cases, and in 2 cases of homozygous Hb E. The results showed
that the amount of Hb Bart’s was increased with the increasing numbers
of the defective α-globin genes. Newborns with single α-globin gene
defect [(α+-thalassemia; -α3.7 or -α4.2)
or Hb Constant Spring and Hb Paksé] have Hb Bart’s at 0.4 ± 0.1% and
0.7 ± 0.1%, respectively. The Hb Bart’s levels found in individuals
with two α-globin gene defects were 2.6 ± 1.2%, 1.6 ± 0.7% and 6.2 ±
2.1% for heterozygous α0-thalassemia (--/αα), homozygotes α+-thalassemia (-α/-α) and compound heterozygous α+-thalassemia/Hb Constant Spring (-α/αTα),
respectively. Newborns with Hb H disease (--/-α) (n=2) had Hb Bart’s at
21.9% and 18.6%, respectively. These results indicated that while Hb
Bart’s detected by CE could be a good marker for α-thalassemia in
newborns, it is not sensitive enough since some cases of α+-thalassemia
carriers had no detectable Hb Bart’s. In contrast, CE is very sensitive
in identifying cases with Hb E in newborns. It could demonstrate Hb E
peak in all 116 cases of Hb E carriers, and variable levels of Hb E
could be measured in all newborns with βE-mutation.
 |
Table 3. Hemoglobin types
and fractions found among 350 newborns in corresponding to genotypes.
Values are presented as mean ± SD or as raw data where
appropriate. |
Discussion
In Thailand, the national thalassemia prevention and control program has been launched formally since 1993.[3]
Two strategies are providing proper treatment of the existing cases and
prevention of new births with three severe thalassemia diseases
including homozygous α0-thalassemia (the Hb Bart’s hydrops fetalis syndrome), homozygous β-thalassemia
and Hb E – β – thalassemia. Step-by-step screening strategy composing
of initial screening using combined red blood cell indices, osmotic
fragility (OF) test and dichlorophenolindophenol (DCIP) test followed
by Hb analysis and DNA testing are applied.[5,6] The
program has been progressed and is active all over the country
including northeast Thailand where our laboratory at Khon Kaen
University has been recognized as one of the reference centers. We have
demonstrated retrospectively on prenatal diagnosis of 756 couples that
the overall proportions of affected fetuses, thalassemia carriers and
unaffected fetuses were respectively 26.9%, 50.0% and 23.1% which were
corresponding quite well with the expected values for a recessive
genetic disorder.[7] This indicates that most of the
targeted thalassemia diseases in the at-risk couples could be
successfully prevented, leading to a substantial reduction in future
cost of treatment of the diseases.
Another approach to
monitoring the performance of a prevention and control program is to
look prospectively at newborns. In this study, we have addressed this
in northeast Thailand after 20 years of a prevention and control
program. Examination for all thalassemic genes found in Thailand was
carried out on 350 cord blood specimens collected consecutively and
prospectively at delivery. As shown in Table 1,
we have noted as many as 22 thalassemia genotypes among 184 of 350
(52.6%) newborns. Moreover, no case with the three severe thalassemia
diseases targeted in a prevention and control program was encountered.
As shown in Table 2, high incidence for all forms of α-thalassemia
(35.8%) and Hb E (39.1%) are detected. In contrast, the frequency of
β-thalassemia is relatively much lower (0.6%). This pattern of
thalassemia and hemoglobinopathies found in newborns is very similar to
those documented in adult population observed in a micro-mapping survey
in the region[8] as well as in other areas of Thailand as shown in Table 2.
Here is indicated effective prevention of new case with severe
thalassemia but the corresponding thalassemic genes are still prevalent
in the region. In fact, this is not unexpected since by theory allele
frequency in the population remains constant from generation to
generation.
Taking the data on Hb analysis into diagnostic
consideration for the three important thalassemia carriers, we found
that Hb Bart’s detected by capillary electrophoresis is a very good
marker for reporting α-thalassemia in newborns. Different levels of Hb
Bart’s were detected for different α-thalassemia genotypes and the
higher level, the more α-globin gene defect (Table 3).
However, as also observed in other studies, Hb Bart’s may be
undetectable in some cases of α-thalassemia especially those with one
α-globin gene defect (-α/αα or αTα/αα).[19,20]
In contrast, we found that all cases with Hb E, either in heterozygote
or homozygote had detectable levels of Hb E on the capillary
electrophoresis system. This simple examination should permit making
initial recognition of the cases before definite diagnosis by DNA
analysis. A problem remains for β-thalassemia. In two cases of β0-thalassemia carriers encountered (αα/αα, β17/βA),
we observed the same Hb FA pattern with that of the normal newborns
i.e. Hb F (92.9 & 86.8 % v.s. 84.2 + 4.5 %) and Hb A (7.1 &
13.0 % v.s. 15.8 + 4.5 %). This confirms that diagnosis of
β-thalassemia is relatively difficult in newborns unless DNA analysis
is performed.[16,17,20,21]
Nonetheless,
our study demonstrates the current prevalence of thalassemia and
hemoglobinopathies among newborns in northeast Thailand after a
prevention and control program of thalassemia has been launched for
more than 20 years. Since the three important thalassemia carriers
including α0-thalassemia,
β-thalassemia, and Hb E are still prevalence in the population, an
effective prevention and control program of thalassemia should be
continuously operated in the region.
References
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia. Hemoglobin 1987; 11: 65-88. https://doi.org/10.3109/03630268709036587 PMid:3294755

- Fucharoen
G, Fucharoen S, Sanchaisuriya K, Sae-ung N, Suyasunanond U, Sriwilai P,
Chinorak P. Frequency distribution and haplotypic heterogeneity of
βE-globin gene among eight minority groups of northeast Thailand. Hum
Hered 2002; 53: 18-22. https://doi.org/10.1159/000048600 PMid:11901267
- Fucharoen
S, Winichagoon P. Thalassemia in Southeast Asia: problems and strategy
for prevention and control. Southeast Asian J Trop Med Public Health
1992; 23: 647-55. PMid:1298071
- Fucharoen
S, Winichagoon P, Thonglairoam V, Siriboon W, Siritanaratkul N,
Kanokpongsakdi S, Vantanasiri C. Prenatal diagnosis of thalassemia and
hemoglobinopathies in Thailand: experience from 100 pregnancies.
Southeast Asian J Trop Med Public Health 1991; 22: 16-29. PMid:1948258
- Fucharoen
G, Sanchaisuriya K, Sae-ung N, Dangwibul S, Fucharoen S. A simplified
screening strategy for thalassemia and hemoglobin E in rural
communities in south-east Asia. Bull World Health Organ 2004; 82:
364-72. PMid:15298227 PMCid:PMC2622836
- Sanchaisuriya
K, Fucharoen S, Fucharoen G, Ratanasiri T, Sanchaisuriya P,
Changtrakul, Y, Ukosanakarn U, Ussawaphark W, Schelp FP. A reliable
screening protocol for thalassemia and hemoglobinopathies in pregnancy;
an alternative approach to electronic blood cell counting. Am J Clin
Pathol 2005; 123: 113–8. https://doi.org/10.1309/FUF9EVGQ24V1PKTP PMid:15762286
- Yamsri
S, Sanchaisuriya K, Fucharoen G, Sae-ung N, Ratanasiri T, Fucharoen S.
Prevention of severe thalassemia in northeast Thailand: 16 years of
experience at a single university center. Prenat Diagn 2010; 30: 540-6.
https://doi.org/10.1002/pd.2514
- Tritipsombut
J, Sanchaisuriya K, Phollarp P, Bouakhasith D, Sanchaisuriya P,
Fucharoen G, Fucharoen S, Schelp FP. Micromapping of thalassemia and
hemoglobinopathies in different regions of northeast Thailand and
Vientaine, Laos People's Democratic Republic. Hemoglobin 2012; 36:
47-56. https://doi.org/10.3109/03630269.2011.637149 PMid:22122810
- Naing
L, Winn T, Rusli BN. Practical issues in calculating the sample size
for prevalence studies. Arch Orofacial Sci 2006; 1: 9-14.
- Srivorakun
H, Fucharoen G, Sae-Ung N, Sanchaisuriya K, Ratanasiri T, Fucharoen S.
Analysis of fetal blood using capillary electrophoresis system: a
simple method for prenatal diagnosis of severe thalassemia diseases.
Eur J Haematol 2009; 83: 57-65. https://doi.org/10.1111/j.1600-0609.2009.01245.x PMid:19226360
- Sae-ung
N, Fucharoen G, Sanchaisuriya K, Fucharoen S. Alpha(0)-thalassemia and
related disorders in northeast Thailand: a molecular and hematological
characterization. Acta Haematol 2007; 117: 78-82. https://doi.org/10.1159/000096857 PMid:17106191
- Singsanan
S, Fucharoen G, Savongsy O, Sanchaisuriya K, Fucharoen S. Molecular
characterization and origins of Hb Constant Spring and Hb Paksé in
Southeast Asian populations. Ann Hematol 2007; 86: 665-9. https://doi.org/10.1007/s00277-007-0310-x PMid:17589844
- Fucharoen
S, Fucharoen G, Sanchaisuriya K, Pengjam Y. Molecular analysis of a
Thai β-thalassaemia heterozygote with normal haemoglobin A2 level:
implication for populations screening. Ann Clin Biochem 2002; 39: 44-9.
https://doi.org/10.1258/0004563021901720 PMid:11853188
- Singha
K, Fucharoen G, Jetsrisuparb A, Fucharoen S. Molecular and
hematological characteristics of a novel form of α-globin gene
triplication: the hemoglobin St.Luke's-Thailand [α95(G2)Pro→Arg] or Hb
St. Luke's [A2] HBA2. Clin Biochem 2013; 46: 675-80. https://doi.org/10.1016/j.clinbiochem.2013.01.022 PMid:23395770
- Yamsri
S, Sanchaisuriya K, Fucharoen G, Sae-ung N, Fucharoen S. Genotype and
phenotype characterizations in a large cohort of β-thalassemia
heterozygote with different forms of α-thalassemia in northeast
Thailand. Blood Cells Mol Dis 2011; 47: 120-4. https://doi.org/10.1016/j.bcmd.2011.05.003 PMid:21664157
- Tritipsombut
J, Sanchaisuriya K, Fucharoen S, Fucharoen G, Siriratmanawong N.
Pinmuang-ngam C, Sanchaisuriya P. Hemoglobin profiles and hematologic
features of thalassemic newborns: application to screening of
alpha-thalassemia 1 and hemoglobin E. Arch Pathol Lab Med 2008; 132:
1739-45. PMid:18976009
- Charoenkwan
P, Taweephol R, Sirichotiyakul S, Tantiprabha W, Sae-Tung R, Suanta S.
Cord blood screening for α-thalassemia and hemoglobin variants by
isoelectric focusing in northern Thai neonates: Correlation with
genotypes and hematologic parameters. Blood Cells Mol Dis 2010; 45:
53-7. https://doi.org/10.1016/j.bcmd.2010.02.015 PMid:20299254
- Munkongdee
T, Pichanun D, Butthep P, Klamchuen S, Chalermpolprapa V, Winichagoon
P, Svasti S, Fucharoen S. Quantitative analysis of Hb Bart's in cord
blood by capillary electrophoresis system. Ann Hematol 2011; 90: 741-6.
https://doi.org/10.1007/s00277-010-1137-4 PMid:21188378
- Rugless
MJ, Fisher CA, Stephens AD, Amos RJ, Mohammed T, Old JM. Hb Bart's in
cord blood: an accurate indicator of alpha-thalassemia. Hemoglobin
2006; 30: 57-62. https://doi.org/10.1080/03630260500454550 PMid:16540417
- Srivorakun
H, Fucharoen G, Changtrakul Y, Komwilaisak P, Fucharoen S. Thalassemia
and hemoglobinopathies in Southeast Asian newborns: diagnostic
assessment using capillary electrophoresis system. Clin Biochem 2011;
44: 406-11. https://doi.org/10.1016/j.clinbiochem.2011.01.006 PMid:21277293
- Uaprasert
N, Settapiboon R, Amornsiriwat S, Sarnthammakul P, Thanapat T,
Rojnuckarin P, Sutcharitchan P. Diagnostic utility of isoelectric
focusing and high performance liquid chromatography in neonatal cord
blood screening for thalassemia and non-sickling hemoglobinopathies.
Clin Chim Acta 2014; 427: 23-6. https://doi.org/10.1016/j.cca.2013.09.041 PMid:24095765
[TOP]