Maimiza Zahari2, Siti Aishah Sulaiman1, Zulhabri Othman1, Yasmin Ayob2, Faraizah Abd Karim2 and Rahman Jamal1.
1 UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia, Jalan Yaacob Latiff, Kuala Lumpur, Malaysia.
2 National Blood Centre, Jalan Tun Razak, Kuala Lumpur, Malaysia.
Correspondence to: Professor Datuk Dr. A Rahman A Jamal. UKM Medical
Molecular Biology Institute (UMBI). Jalan Yaacob Latif, Bandar Tun
Razak, 56000 Cheras, Kuala Lumpur, Malaysia. Tel: +60391459000, Fax:
+60391717185. E-mail:
rahmanj@ppukm.ukm.edu.my
Published: September 1, 2018
Received: May 17, 2018
Accepted: August 10, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018056 DOI
10.4084/MJHID.2018.056
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background.
Haemophilia A (HA) and Haemophilia B (HB) are X-linked blood disorders
that are caused by various mutations in the factor VIII (F8) and factor IX (F9)
genes respectively. Identification of mutations is essential as some of
the mutations are associated with the development of inhibitors. This
study is the first comprehensive study of the F8 mutational profile in Malaysia.
Materials and methods. We analysed 100 unrelated HA and 15 unrelated HB patients for genetic alterations in the F8 and F9
genes by using the long-range PCR, DNA sequencing, and the
multiplex-ligation-dependent probe amplification assays. The prediction
software was used to confirm the effects of these mutations on factor
VIII and IX proteins.
Results. 44 (53%) of the severe HA patients were positive for F8 intron 22 inversion, and three (3.6%) were positive for intron one inversion. There were 22 novel mutations in F8,
including missense (8), frameshift (9), splice site (3), large deletion
(1) and nonsense (1) mutations. In HB patients, four novel mutations
were identified including the splice site (1), small deletion (1),
large deletion (1) and missense (1) mutation.
Discussion. The mutational spectrum of F8
in Malaysian patients is heterogeneous, with a slightly higher
frequency of intron 22 inversion in these severe HA patients when
compared to other Asian populations. Identification of these mutational
profiles in F8 and F9
genes among Malaysian patients will provide a useful reference for the
early detection and diagnosis of HA and HB in the Malaysian population.
|
Introduction
Haemophilia is an inherited X-linked blood disorder which causes prolonged bleeding time after injuries or trauma.[1] Haemophilia A (HA) and Haemophilia B (HB) are due to the deficiency of coagulation factor VIII (gene, F8) and factor IX (gene, F9) respectively.[2]
Upon activation, factor VIII and factor IX form an active complex
(tenase complex) which activates factor X and the following factors in
the coagulation pathway.[1] Thus, a deficiency or
dysfunction of any of these factors can impair clot formation and
consequently causes bleeding diathesis.
In HA cases, the most
recurrent genetic mutations are the inversion of intron 22 (IVS22),
which accounts for about 45% of severe patients,[3] and intron 1 (IVS1) which accounts for 2-5% of severe patients.[4] As for HB patients, the most common mutations identified in the F9 gene are the missense mutations (74%).[5] Apart from these mutations, there is a wide range of different genetic alterations spread throughout F8 and F9 genes, including single nucleotide substitutions, small and large deletions.[5-7] Until now, about 1968 unique variants of F8 are listed in the factor VIII database,[6,7] and 1094 unique variants of F9 in the factor IX database.[5]
The
current standard of treatment of HA and HB is primary prophylaxis, with
regular infusion of factor VIII or factor IX respectively to prevent
joint bleeding and damage.[8] However, the development of inhibitors in these patients is a severe complication of this infusion therapy.[9] Such inhibitory response happens in 25-30% of HA patients[10,11] and 1-4% of HB patients,[12,13] and these incidences may be higher depending on the ethnicity.[14,15] A
systematic review showed that factor VIII inhibitory response was
strongly associated with large deletions and nonsense mutations
compared to IVS22 mutation,[16] therefore suggesting
a strong genetic predisposition, hence the importance of identifying
such mutations before commencing the infusion treatment. In Malaysia,
despite the prevalence of HA and HB are around 5.9/100000 males and
1.0/100000 males respectively,[17] there have been only small studies on the mutational status of F8 (only in exon 14)[18] and in F9[19,20] genes. Therefore, this study aimed to investigate comprehensively the mutational spectrums of F8 and F9
genes in a representative cohort of Malaysian patients corresponding to
their disease severity as well as the inhibitory response.
Materials and Methods
Sample collection.
This study was approved by the Universiti Kebangsaan Malaysia Ethics
Committee and the ethics committee of the Ministry of Health of
Malaysia. Written informed consent taken from all patients with
confirmed non-familial HA (n=100) and HB (n=15) who were being
followed-up at the National Blood Centre, Kuala Lumpur. Detailed
clinical history along with pedigree data were taken, and the disease
severity classification was as the following: 1) mild HA
(FVIII/FIX:C:>5-40%), 2) moderate HA (FVIII/FIX:C:1-5%), and 3)
severe HA (FVIII/FIX:C:<1%).[21] Venous blood
(10mL) was collected in EDTA Vacutainer collection tubes (BD, New
Jersey, USA) and proceeded to DNA extraction using the salting-out
extraction method.[22] DNA quality and concentration
were determined by using NanoDrop Spectrophotometry and gel
electrophoresis according to the manufacturer's instruction.
Detection of intron 22 inversion (IVS22) in F8. Detection of IVS22 in F8 performed by using the Long-Range PCR kit (QIAGEN, Hilden, USA) according to previously published methods[23,24]
with slight modifications. Primers P, Q, A & B (Life Technologies,
Wien, Austria) were utilised to amplify the region of interests.[23,24]
Briefly, a total of 25 µl PCR reaction which contained 20ng genomic
DNA, LD-PCR reaction master-mix (Qiagen LD-PCR kit, Hilden, USA), 7.5%
DMSO, 10 mM of 7-deaza-dGTP and 10 pmol of primers P, Q, A & B in
each single-tube PCR reaction. Conditions for the PCR reaction: initial
denaturation at 95 °C for 2 min and 15 s; followed by 30 cycles of
denaturation at 95 °C for 12 s, annealing at 65 °C for 30 s and
elongation at 68 °C for 12 min for the first ten cycles. The remaining
20 cycles had 20 s addition to each cycle step. Confirmation of the PCR
products was visualised using agarose gel electrophoresis.
Detection of intron 1 inversion (IVS1) in F8. Detection of IVS1 in F8 done by using the PCR Core kit (Roche Diagnostics, Indiana, USA) according to the previously published methods and primers.[4] The PCR products were visualised using agarose gel electrophoresis.
Detections of other mutations in F8 and F9. F8
coding regions (26 exons) including the intron/exon boundaries and the
promoter regions were amplified using 26 sets of previously published
primers and methods.[25] The entire F9
coding region (8 exons) including the intron/exon boundaries, the
promoter region, and the polyadenylation site was amplified using eight
sets of previously published primers and methods.[26,27]
The confirmation of the PCR products was performed using gel
electrophoresis and sequenced using the BigDye Terminator v3.1
sequencing kit (Applied Biosystems, California, USA) on an ABI 3130xl
Genetic Analyzer (Applied Biosystems, California, USA) according to the
manufacturer’s instructions.
Detection of large deletions in F8 and F9.
Samples that did not show any exon amplification (but flanking exon
amplification) or did not show any mutations were suspected of having
large deletions. Detection of large deleted regions in F8 and F9 was performed using the multiplex-ligation-independent probe amplification (MLPA) kits, namely the SALSA MLPA P178 F8 and SALSA MLPA P207-C1 F9
probe mix kits (MRC-Holland, Amsterdam, Netherlands), according to the
manufacturer’s instructions. Amplified PCR products were separated by
ABI 3130xl Genetic Analyzer (Applied Biosystems, California, USA) with
LIZ-500 (Applied Biosystems, California, USA) as the size standard. The
data was analysed using Coffalyser.Net (MRC-Holland, Amsterdam,
Netherlands) according to the provided guidelines. Probe ratios <0.7
were considered as deletions and probe ratios >1.3 as duplications.
Negative controls were the donor DNA samples from healthy males.
Molecular genetic analysis and nomenclature. F8 and F9
nucleotide numbering (c.) is designated according to coding bases from
A (nucleotide+1) from the initiation codon for methionine (ATG) at
position -171 (F8:ref. NM_000132.3) and (ATG) at position -29 (F9:ref.
NM_000133.3) respectively. While the protein numbering (p.) follows the
amino acid sequences that assign the first residue methionine as +1 in
each factor VIII and IX sequences (FVIII: NP_000123.1 and FIX:
NP_000124.1 respectively) according to the Human Genome Variation
Society guidelines.[28] Sequence variants were
aligned with the corresponding wild-type sequences using BLAST (NCBI)
and compared to the HA and HB mutation databases (Factor 8,[6,7] Factor 9,[5] Human Gene Mutation Database and CDC Haemophilia A Mutation Project database[29]).
Novel variants further analysed for their effects on the factor VIII
and IX protein by using multiple software, including Sorting Intolerant
From Tolerant (SIFT) and Polymorphism Phenotyping (PolyPhen2),[30,31] PROVEAN (Protein Variation Effect Analyzer)[32] and Mutation Taster2.[33]
A SIFT score ranges from deleterious (< or equal to 0.05) to
tolerated SNP (> 0.05). For PolyPhen2, the score ranges from 0.0
(tolerated) to 1.0 (deleterious). The PROVEAN score of an equal to or
less than a predefined threshold of -2.5 value indicates for a
"deleterious" effect. The visualisation of affected amino acid was
performed on a crystal structure of the protein from the Protein Data
Bank database for factor VIII protein (PDB-ID:2R7E)[34] and IX protein (PDB-ID:2WPI)[35] using Pymol, version 1.8.6.1 that is freely available online.
Results
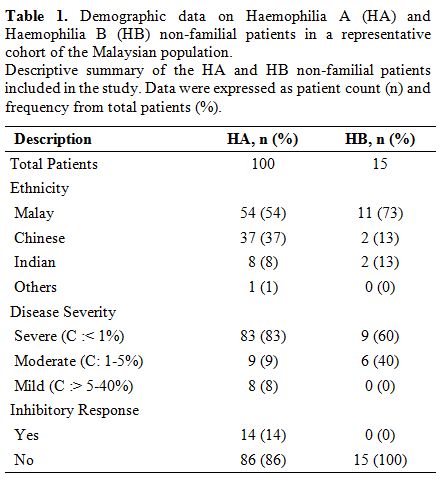
Demographic and clinical data.
Among the 100 HA patients in the study, 83 were severe
(FVIII:C:<1%), nine were moderate (FVIII:C:1-5%), and eight were
mild (FVIII:C:>5-40%) whereas, for the 15 HB patients, nine were
severe (FIX:C:<1%), and six were moderate (FIX:C:1-5%) (Table 1).
Fourteen of the severe HA patients developed inhibitors against factor
VIII, while none of the HB patients had factor IX inhibitor. Majority
of the patients were Malays (HA:54%, HB:73%), and followed by Chinese
(HA:37%, HB:13%), Indians (HA:8%, HB:13%) and other (HA:1%) (Table 1).
 |
Table
1. Demographic data on Haemophilia A (HA) and Haemophilia B (HB)
non-familial patients in a representative cohort of the Malaysian
population.
Descriptive summary of the HA and HB non-familial
patients included in the study. Data were expressed as patient count
(n) and frequency from total patients (%).
|
F8 mutations.
Out of 83 severe HA patients, 44 (53%) of them were positive for intron
22 inversion (IVS22), and three (3.6%) were positive for intron 1
inversion (IVS1). Among those 44 IVS22 positive patients, 18 of them
had sporadic occurrence while 26 were familial based on the family
history. For those remaining HA patients without IVS22/1 mutations, a
total of 22 novel mutations were identified (Table 2)
consisting of missense (8), frameshift (9), splice site (3), large
deletion (1) and nonsense (1) mutations. Additionally, 21 of previously
reported F8 mutations were also detected (Table 2)
consisting large deletions (2), missense (10), nonsense (2), splice
site (4) and frameshift (3) mutations. Excluding the IVS22 and IVS1
mutations, 41.7% of these identified mutations mainly occurred at the
exon 14 of F8. In three
severe HA patients, three large deletions were detected. Patient HA93
has a novel deletion which spans from exon 8 to exon 12 corresponding
to A1-a1-A2 domains of factor VIII. Whereas, patient HA1 and HA41 have
large deletions that span from exon 4 to exon 6 (corresponding to the
A1 domain of factor VIII) and span from exon 8 to exon 9 (corresponding
to A1-a1-A2 domains of factor VIII) respectively (Table 2). Unfortunately, we were unable to detect any mutation in four of the severe HA patients (HA4, HA15, HA60, and HA64).
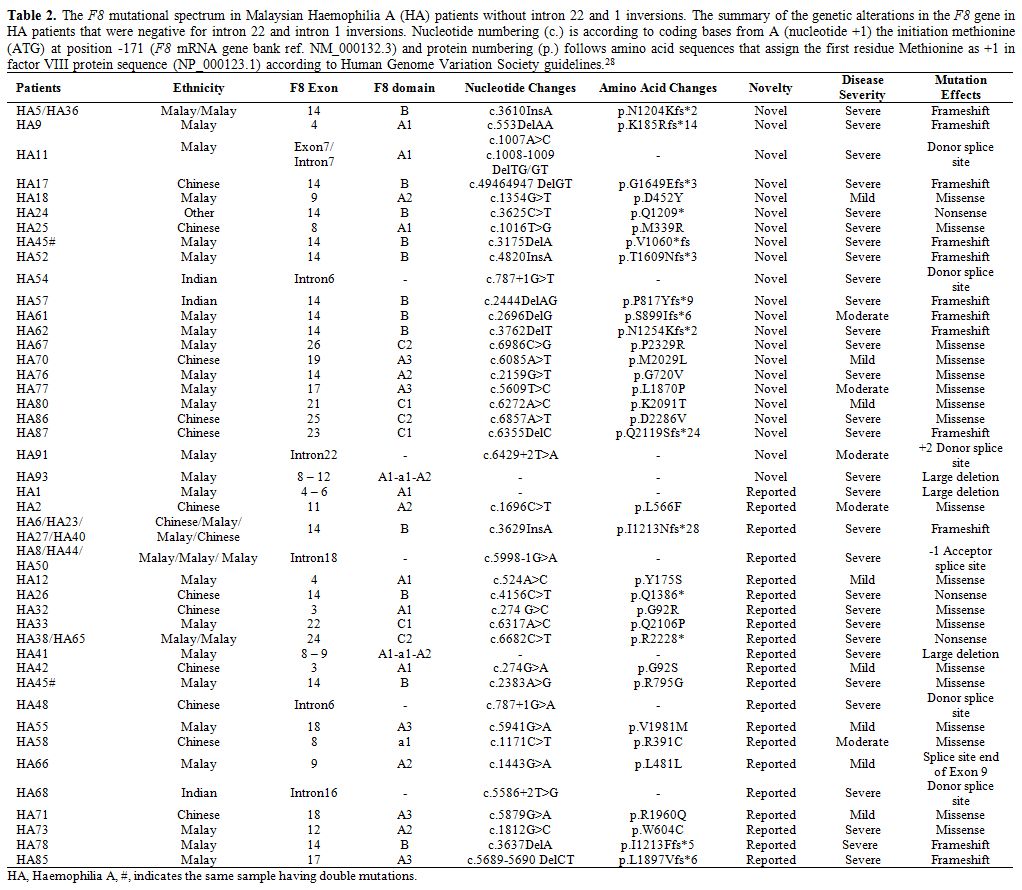 |
Table 2. The F8
mutational spectrum in Malaysian Haemophilia A (HA) patients without
intron 22 and 1 inversions. The summary of the genetic alterations in
the F8 gene in HA patients
that were negative for intron 22 and intron 1 inversions. Nucleotide
numbering (c.) is according to coding bases from A (nucleotide +1) the
initiation methionine (ATG) at position -171 (F8
mRNA gene bank ref. NM_000132.3) and protein numbering (p.) follows
amino acid sequences that assign the first residue Methionine as +1 in
factor VIII protein sequence (NP_000123.1) according to Human Genome
Variation Society guidelines.[28] |
To
further evaluate the impact of the novel missense mutations identified,
we performed prediction analysis on the effect of these mutations on
the factor VIII protein using multiple prediction software (Table 3).
Except for one missense mutation in a mild HA patient, all other novel
missense mutations were predicted to have damaging effects (Table 3). We also visualised the amino acid location of these novel missense mutations in factor VIII (Figure 1).
For example, the HA67 patient who has a severe disease was detected to
have a missense mutation of c.6986C>G that results in the
substitution of proline to arginine at position 2329, and this mutation
was predicted to be damaging. In the wildtype position, the large
cyclic hydrophobic residue of Pro2329 lies within the C2 domain and
forms hydrogen bonds with the neighbouring polar residue of glutamine (Figure 1B).
Substitution of this hydrophobic proline to the positively charged
arginine would disrupt this hydrogen bond. Patient HA77 who has a
moderate disease was detected to have a missense mutation of
c.5609T>C, which was also predicted to be damaging. For this
mutation, the wildtype residue of Leu1870 forms a hydrogen bond with
the polar Ser1819 residue through its hydroxyl group in the A3 domain (Figure 1C).
Substitution of Leu1870 to cyclic proline would alter the hydroxyl
group interaction therefore consistent with the predicted damaging
score. Patient HA70 who has a mild disease also has a non-damaging
missense mutation of c.6085A>T. For this mutation, the wildtype
residue of Met2029 forms a hydrogen bond with the positively charged
histidine residue in the A3 domain near to the core (Figure 1D).
Substitution of this methionine to leucine that is similar in structure
and polarity would minimally disrupt this bond, and thus consistent
with the predicted score.
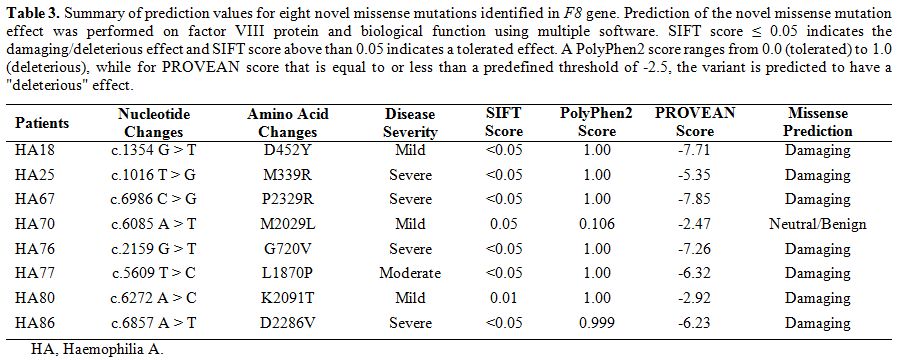 |
Table 3.
Summary of prediction values for eight novel missense mutations identified in F8
gene. Prediction of the novel missense mutation effect was performed on
factor VIII protein and biological function using multiple software.
SIFT score ≤ 0.05 indicates the damaging/deleterious effect and SIFT
score above than 0.05 indicates a tolerated effect. A PolyPhen2 score
ranges from 0.0 (tolerated) to 1.0 (deleterious), while for PROVEAN
score that is equal to or less than a predefined threshold of -2.5, the
variant is predicted to have a "deleterious" effect. |
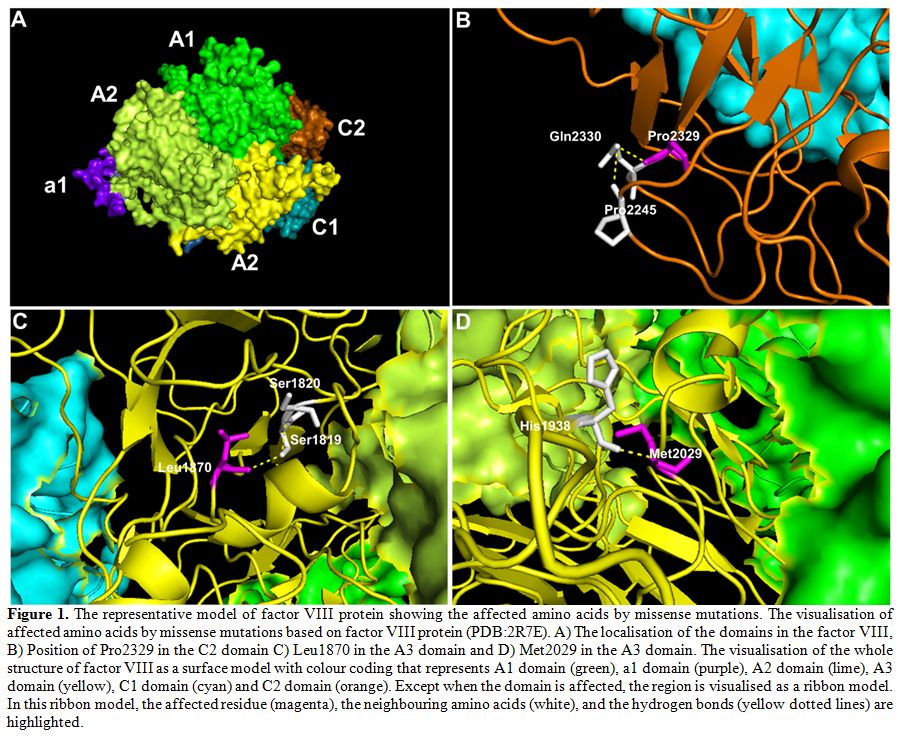 |
Figure 1. The
representative model of factor VIII protein showing the affected amino
acids by missense mutations. The visualisation of affected amino acids
by missense mutations based on factor VIII protein (PDB:2R7E). A) The
localisation of the domains in the factor VIII, B) Position of Pro2329
in the C2 domain C) Leu1870 in the A3 domain and D) Met2029 in the A3
domain. The visualisation of the whole structure of factor VIII as a
surface model with colour coding that represents A1 domain (green), a1
domain (purple), A2 domain (lime), A3 domain (yellow), C1 domain (cyan)
and C2 domain (orange). Except when the domain is affected, the region
is visualised as a ribbon model. In this ribbon model, the affected
residue (magenta), the neighbouring amino acids (white), and the
hydrogen bonds (yellow dotted lines) are highlighted. |
F9 mutations. From the 15 HB patients, there were four novel mutations identified (Table 4),
namely the splice site mutation (1), small deletion (1) and large
deletion (1) and missense (1) mutations. A novel large deletion spans
from exon 1 to exon 4, corresponding to signal-propeptide-GLA-EGF1
domains of factor IX, was detected in patient HB13 who has a severe
disease. Additionally, there were ten previously reported mutations in
the F9 identified among the patients (Table 4)
consists of the splice site (2), missense (5), nonsense (2) and
frameshift (1) mutations. A novel missense mutation (c.803G>A) in
the patient HB4 was predicted to be damaging (SIFT score= <0.05,
PolyPhen2 score= 1.00, PROVEAN score= -10.52). This missense mutation
(c.803G>A) resulted in the substitution of cysteine to tyrosine
residue at position 268. In this serine-peptidase catalytic domain, a
wildtype residue of Cys268 forms strong disulphide hydrogen bonds with
small hydrophobic Ala266 residue, contributing to the helical structure
and folding of the factor IX (Figure 2B). Substitution of this Cys268 residue with tyrosine would affect the structure of the protein.
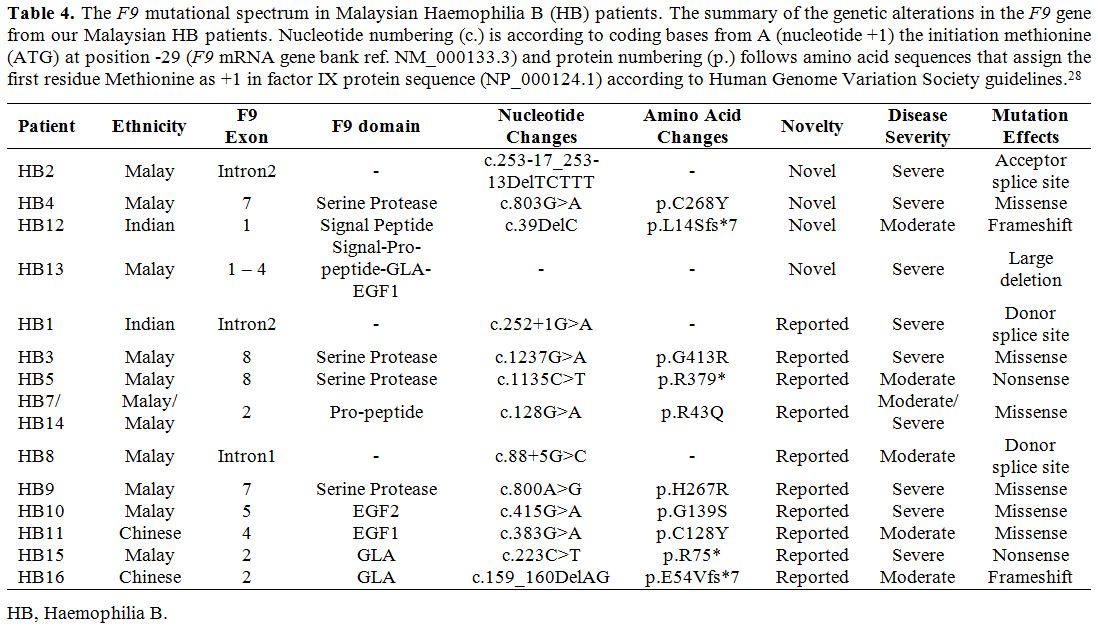 |
Table 4.
The F9 mutational spectrum in Malaysian Haemophilia B (HB) patients. The summary of the genetic alterations in the F9 gene
from our Malaysian HB patients. Nucleotide numbering (c.) is according
to coding bases from A (nucleotide +1) the initiation methionine (ATG)
at position -29 (F9 mRNA gene
bank ref. NM_000133.3) and protein numbering (p.) follows amino acid
sequences that assign the first residue Methionine as +1 in factor IX
protein sequence (NP_000124.1) according to Human Genome Variation
Society guidelines.[28] |
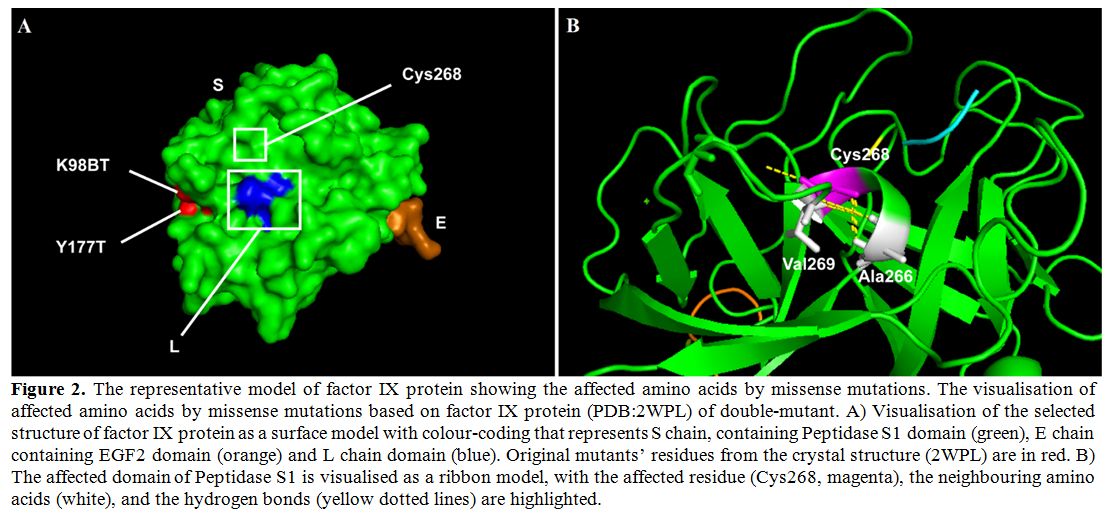 |
Figure 2. The
representative model of factor IX protein showing the affected amino
acids by missense mutations. The visualisation of affected amino acids
by missense mutations based on factor IX protein (PDB:2WPL) of
double-mutant. A) Visualisation of the selected structure of factor IX
protein as a surface model with colour-coding that represents S chain,
containing Peptidase S1 domain (green), E chain containing EGF2 domain
(orange) and L chain domain (blue). Original mutants’ residues from the
crystal structure (2WPL) are in red. B) The affected domain of
Peptidase S1 is visualised as a ribbon model, with the affected residue
(Cys268, magenta), the neighbouring amino acids (white), and the
hydrogen bonds (yellow dotted lines) are highlighted. |
Genotype-Phenotype relationship in HA and HB patients.
From those 44 severe HA patients with the IVS22 mutation, five of them
developed inhibitors against factor VIII, while two of the three severe
HA patients with IVS1 mutation also had factor VIII inhibitors (Table 5).
Among those remaining patients negative for IVS22/1 mutations, seven
patients had inhibitory presence with different types of mutations,
namely patient HA38 (nonsense mutation), patient HA73 (missense
mutation) and patient HA4 (undetected mutation), patients HA85 and HA87
(small deletions) and patients, HA41 and HA93 (large deletions). In
contrast, none of the HB patients exhibited any inhibitory response
against factor IX.
 |
Table 5. Factor VIII inhibitory response distribution across the mutational spectrum of F8
in a representative cohort of Malaysians. Factor VIII inhibitory
response distribution across the mutation types in Haemophilia A
patients (n=14) within the F8
gene. Data were expressed as count (n) and frequency (%). Numbers of
mutations are not equal to the number of patients, due to some patients
share the same mutations. IVS, Intron Inversion. |
Discussion
This study is the first to report a comprehensive mutational spectrum of F8 in 100 HA patients from Malaysia, together with the mutational profile of F9
in 15 HB patients. Among the 83 severe and non-familial HA patients,
53% of them have IVS22 mutation which is slightly higher than other
Asian populations.[36-40] We also identified 22 novel mutations in F8 and four novel mutations in F9. Among those with novel mutations, we found one HA patient and one HB patient each with a novel large deletion in the F8 and F9 respectively, and both patients have severe disease. Unfortunately, we were unable to detect any mutation in F8
in four HA patients with severe disease. Similarly, previous studies
also reported that in a small number of HA patients, no mutation was
detected despite the use of multiple techniques.[38,41,42] This is probably due to the location of the mutations which lie deep within the introns or outside the analysable region of F8 using the current techniques.[43,44] Given the associations between F8 and F9
mutational status and the disease severity as well as the development
of inhibitors following the treatment, our findings suggest that
detection of the mutational spectrum of F8 and F9 can improve the disease management and treatment outcome in HA and HB patients in Malaysia.
Three
of the severe HA patients have large deletions, including one novel
deletion. Patient HA93 with the novel deletion affecting A1-a1-A2
domains of factor VIII showed a high inhibitor level at the age of 12
years. Likewise, patient HA41 with a large deletion of A1-a1-A2 domains
also had a high inhibitor level since the age of 4 years. Large
deletions and nonsense mutations identified in factor VIII are shown to
associate with higher risk of inhibitory response,[16,36,45-47] particularly at the A2 and C2 domains.[48]
This evidence is in agreement with our findings, as our patient HA1 who
has a large deletion affecting only the A1 domain did not develop the
inhibitory response. Thus, depending on which factor VIII domains that
are being affected by the large deletions, a differential outcome in
the inhibitory response may be observed. Therefore, further
investigations are needed to evaluate the prognostic value of these
large deletions in predicting the inhibitory response.
Excluding
IVS22 and IVS1 mutations, 41.7% of the identified mutations in the
present study mainly occurred in exon 14 that is corresponding to the B
domain of factor VIII. B domain has no homology sequence to any other
known genes and has been shown to participate in the intracellular
processing and trafficking of factor VIII.[49] The role of the B domain in the pro-coagulant activity is minimal, as this domain is cleaved off during the activation.[50,51]
Here, we reported that among our eight severe HA patients, seven novel
nonsense/frameshift mutations identified were in exon 14. Previous
studies have reported that only some of these nonsense/frameshift
mutations were causative mutations,[52] despite that
they could result in premature termination or frameshift codon.
Therefore, further works should be pursued to elucidate the pathogenic
impact of these mutations on factor VIII activity and production. Due
to the limitation of the budget, we did not perform a functional study
to assess the effects of these novel exon 14 mutations. However, four
of these identified mutations are near to the ‘hotspot’ region at codon
position 1210-1213 that is associated with a severe phenotype.[53] Further functional studies are needed to elucidate the mechanism of how these mutations can affect factor VIII activity.
Missense mutations may represent polymorphisms,[54] thus may require further evaluation. In the present study, seven novel missense mutations in F8 were predicted to be damaging. The structural visualisations of the affected amino acids in factor VIII (Figure 1)
were consistent with the prediction scores, therefore suggesting that
these mutations are more likely to be disease-causing. For example, we
identified that patient HA67 has the strongest prediction of
disease-causing mutation (Pro2326Arg) in the C2 domain, and this was
consistent with the severe disease phenotype exhibited by the patient.
Any mutation that lies within the C2 domain is likely to be causative
as the C1/2 domains are essential for the binding of factor VIII to the
von Willebrand factor[55] and membrane-binding motif of tenase complex.[56,57]
In comparison to previous studies, we found 21 recurrent mutations in F8. Among them, a nonsense mutation (c.6682C>T) was reported in various populations,[6,7] including two cases from the Asian populations.[58,59] Another recurrent missense mutation (c.1171C>T, Arg391Cys) was also reported before with differential disease outcomes,[6,7] even though this mutation lies within the thrombin activation site.[60-62]
HA disease severity can vary upon which substitution of the amino acid
at Arg391/Arg372 (mature protein) that can influence the rate of
thrombin cleavage.[63] Histidine residue substitution
at Arg372 position resulted in lower activation and thrombin cleavage,
though no effect on factor VIII pro-coagulation activity,[63]
thus consistent with the mild disease phenotype. Whereas, Arg372
substitutions to cysteine, leucine, and proline residues were reported
in moderate to severe HA patients,[6,7] due to impairments in thrombin cleavage and activity.[64,65] Interestingly, our severe patient HA45 has double mutations in F8,
namely one novel frameshift deletion (c.3175DelA) and a previously
reported missense mutation (c.2383A>G) in one Taiwanese woman with a
severe disease phenotype.[66] As this recurrent
missense mutation (c.2383A>G) lies within the B domain, therefore,
it may not be a disease-causing mutation. However, a presence of
frameshift deletion would affect the factor VIII synthesis and
function.
We identified four novel mutations in F9,
including in the severe patient HB13 who had a novel large deletion
affecting the signal-pro-peptide-GLA-EGF1 domains of factor IX. This
finding is consistent with the previous findings that 90% of the large
deletions identified were in severe HB patients[48,51,60] and associated with higher risk of inhibitor development.[67]
One HB2 patient with severe disease has a novel small 5bp deletion
(c.253-17_253-13delTCTTT) at the acceptor splice site in intron 2. This
novel small deletion is similar to a previously reported 5bp deletion
(c.253-18_253-14delTTCTT) in two Malaysian siblings with moderate
disease.[19] Despite a difference of a single nucleotide position, the two siblings[19]
and our patient exhibited differential disease outcomes, in which our
5bp deletion is more detrimental due to being nearer to the intron-exon
boundary. As this novel 5bp deletion may interfere with the
acceptor-binding site and causing an exon skipping event,[68]
therefore it could explain such differential disease phenotypes. We
also found a novel frameshift mutation in the signal-peptide domain
(patient HB12) in which this deletion of C nucleotide is consistent
with previous findings that any mutation lies within the early pre-pro
leader sequences of factor IX is detrimental.[69] In
our severe HB4 patient, the novel missense mutation (c.803G>A) is
likely a disease-causing mutation as it lays within the serine-protease
domain and is also predicted to disrupt the helix structure of factor
IX (Figure 2B), consistent with the vital role of the serine-protease domain in factor IX activity.[33]
In comparison to previous studies of F9,
we found ten previously reported mutations. Both nonsense mutations
(c.1135C>T and c.223C>T) have been reported in various
populations,[5] including two Malaysian patients (c.1135C>T only).[19]
Similarly, a frameshift mutation (c.159_160DelAG), the missense
mutations (c.415G>A and c.128G>A) and a splice site mutation
c.252+1G>A were also reported before in Malaysian patients.[19]
As for the remaining recurrent missense mutations, these have been
reported in non-Malaysian populations. The missense mutation of
c.383G>A was reported before in German[70] and Indian patients[71]
though, the latter had a severe disease phenotype. The recurrence
splice site mutation, c.88+5G>A was reported in a Chinese patient
with the same moderate disease.[72] Two recurrence
missense mutations (c.1237G>A and c.800A>G) in our patients are
possibly the disease-causing missense mutations because they are within
the serine-peptidase domain.[73] Intriguingly, a
missense mutation (c.800A>G) in our severe HB9 patient was reported
before with differential disease phenotypes across two different
populations namely, in a French patient with a moderate phenotype[74] and an Indian patient with a severe phenotype.[75]Conclusion
This study is the first to comprehensively analyse the mutational spectrum of F8
in HA patients in Malaysia. The 53% prevalence of the IVS22 mutation in
our severe HA patients is slightly higher than other Asian populations.
A total of 22 and four novel mutations were identified in F8 and F9
respectively, thus suggesting a high heterogeneity of molecular changes
in factor VIII and IX in our local patients. How these mutations can
affect the disease severity and the inhibitor development, is worth
exploring further to provide a better understanding of the
genotype-phenotype association in our patients. These mutational
profiles of our Malaysian HA and HB patients can provide a useful
reference database in the detection of carrier status and the diagnosis
of HA and HB in the Malaysian population.Acknowledgements
The authors would
like to acknowledge the Haematology Department of Singapore General
Hospital for generously sharing their procedures of PCR amplification
and direct sequencing.
References
- Bowen DJ. Haemophilia A and haemophilia B: Molecular insights. Mol Pathol 2002;55(2):127-44. https://doi.org/10.1136/mp.55.2.127 PMid:11950963 PMCid:PMC1187163

- de
Brasi C, El-Maarri O, Perry DJ, Oldenburg J, Pezeshkpoor B, Goodeve A.
Genetic testing in bleeding disorders. Haemophilia 2014;20(0 4):54-8. https://doi.org/10.1111/hae.12409
- Lakich
D, Kazazian HH, Antonarakis SE, Gitschier J. Inversions disrupting the
factor VIII gene are a common cause of severe haemophilia A. Nat Genet
1993;5(3):236-41. https://doi.org/10.1038/ng1193-236 PMid:8275087
- Bagnall
RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking
intron 1 of the factor VIII gene is a frequent cause of severe
hemophilia A. Blood 2002;99(1):168-74. 10.1182/blood.V99.1.168 https://doi.org/10.1182/blood.V99.1.168
- Rallapalli
PM, Kemball-Cook G, Tuddenham EG, Gomez K, Perkins SJ. An interactive
mutation database for human coagulation factor IX provides novel
insights into the phenotypes and genetics of hemophilia B. J Thromb
Haemost 2013;11(7):1329-40. 10.1111/jth.12276 https://doi.org/10.1111/jth.12276
- EAHAD Coagulation Factor Variant Databases. 2017 [cited 13th March 2017]. Available from: http://www.factorviii-db.org/index.php.
- Factor 8 Variant Database. 2014. Available from: http://factorviii-db.org/.
- Franchini M. The modern treatment of haemophilia: a narrative review. Blood Transfusion 2013;11(2):178-82. 10.2450/2012.0166-11
- Walsh
CE, Soucie JM, Miller CH, and the United States Hemophilia Treatment
Center N. Impact of inhibitors on hemophilia a mortality in the United
States. Am J Hematol 2015;90(5):400-5. 10.1002/ajh.23957 https://doi.org/10.1002/ajh.23957
- Wight
J, Paisley S. The epidemiology of inhibitors in haemophilia A: A
systematic review. Haemophilia 2003;9(4):418-35.
10.1046/j.1365-2516.2003.00780.x https://doi.org/10.1046/j.1365-2516.2003.00780.x
- Lacroix-Desmazes
S, Scott DW, Goudemand J, van den Berg M, Makris M, van Velzen AS,
Santagostino E, Lillicrap D, Rosendaal FR, Hilger A, Sauna ZE,
Oldenburg J, Mantovani L, Mancuso ME, Kessler C, Hay CRM, Knoebl P, Di
Minno G, Hoots K, Bok A, Brooker M, Buoso E, Mannucci PM, Peyvandi F.
Summary report of the First International Conference on inhibitors in
haemophilia A. Blood Transfusion 2017;15(6):568-76.
10.2450/2016.0252-16
- Kamiya
T, Takahashi I, Saito H. Retrospective study of inhibitor formation in
Japanese hemophiliacs. Int J Hematol 1995;62(3):175-81. https://doi.org/10.1016/0925-5710(95)00405-H
- Ljung R. Gene mutations and inhibitor formation in patients with hemophilia B. Acta Haematol 1995;94(Suppl. 1):49-52. https://doi.org/10.1159/000204029 PMid:7571995
- Aledort
LM, Dimichele DM. Inhibitors occur more frequently in African-American
and Latino haemophiliacs. Haemophilia 1998;4(1):68. https://doi.org/10.1046/j.1365-2516.1998.0146c.x
- Carpenter
SL, Michael Soucie J, Sterner S, Presley R, Hemophilia Treatment Center
Network I. Increased prevalence of inhibitors in Hispanic patients with
severe haemophilia A enrolled in the Universal Data Collection
database. Haemophilia 2012;18(3):e260-e5.
10.1111/j.1365-2516.2011.02739.x https://doi.org/10.1111/j.1365-2516.2011.02739.x
- Gouw
SC, van den Berg HM, Oldenburg J, Astermark J, de Groot PG, Margaglione
M, Thompson AR, van Heerde W, Boekhorst J, Miller CH, le Cessie S, van
der Bom JG. F8 gene mutation type and inhibitor development in patients
with severe hemophilia A: Systematic review and meta-analysis. Blood
2012;119(12):2922-34. 10.1182/blood-2011-09-379453 https://doi.org/10.1182/blood-2011-09-379453
- Malaysian
Ministry of Health M. Health technology assessment report: Management
of haemophilia. Kuala Lumpur, Malaysia: Ministry of Health, 2012
Contract No.: MOH/P/PAK/258.12(TR).
- Moses
EJ, Ling SP, Al-Hassan FM, Karim FA, Yusoff NM. Identification of novel
mutations in exon 14 of the F8 gene in Malaysian patients with severe
Hemophilia A. Indian J Clin Biochem 2012;27(2):207-8.
10.1007/s12291-011-0161-z https://doi.org/10.1007/s12291-011-0161-z
- Balraj
P, Ahmad M, Khoo AS, Ayob Y. Factor IX mutations in haemophilia B
patients in Malaysia: a preliminary study. Malays J Pathol
2012;34(1):67-9. PMid:22870602
- Ishak
R, Zakaria Z. Detection of carrier status of hemophilia B using DNA
markers. Southeast Asian J Trop Med Public Health 1997;28(3):629-30.
PMid:9561621
- White
GC, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J, on
behalf of the Factor VIII and Factor IX Subcommittee. Definitions in
Hemophilia. Recommendation of the scientific subcommittee on factor
VIII and factor IX of the scientific and standardization committee of
the International Society on thrombosis and haemostasis. Thromb Haemost
2001;85(3):560-. https://doi.org/10.1055/s-0037-1615621 PMid:11307831
- Miller
SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting
DNA from human nucleated cells. Nucleic Acids Res 1988;16(3):1215. https://doi.org/10.1093/nar/16.3.1215 PMid:3344216 PMCid:PMC334765
- Liu
Q, Nozari G, Sommer SS. Single-tube polymerase chain reaction for rapid
diagnosis of the inversion hotspot of mutation in Hemophilia A. Blood
1998;92(4):1458-9. PMid:9694739
- Liu
Q, Sommer SS. Subcycling-PCR for multiplex long-distance amplification
of regions with high and low GC content: Application to the inversion
hotspot in the factor VIII gene. Biotechniques 1998;25(6):1022-8. https://doi.org/10.2144/98256rr01 PMid:9863056
- Vidal
F, Farssac E, Altisent C, Puig L, Gallardo D. Rapid Hemophilia A
molecular diagnosis by a simple DNA sequencing procedure:
Identification of 14 novel mutations. Thromb Haemost 2001;85(4):580-3. https://doi.org/10.1055/s-0037-1615637 PMid:11341489
- Hinks
JL, Winship PR, Makris M, Preston FE, Peake IR, Goodeve AC. A rapid
method for haemophilia B mutation detection using conformation
sensitive gel electrophoresis. Br J Haematol 1999;104(4):915-8.
10.1046/j.1365-2141.1999.01274.x https://doi.org/10.1046/j.1365-2141.1999.01274.x
- Vidal
F, Farssac E, Altisent C, Puig L, Gallardo D. Factor IX gene sequencing
by a simple and sensitive 15-hour procedure for haemophilia B
diagnosis: Identification of two novel mutations. Br J Haematol
2000;111(2):549-51. 10.1111/j.1365-2141.2000.02389.x https://doi.org/10.1111/j.1365-2141.2000.02389.x
- den
Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS,
McGowan-Jordan J, Roux A-F, Smith T, Antonarakis SE, Taschner PEM, on
behalf of the Human Genome Variation Society, the Human Variome
Project, and the Human Genome Organisation. HGVS Recommendations for
the description of sequence variants: 2016 Update. Hum Mutat
2016;37(6):564-9. 10.1002/humu.22981 https://doi.org/10.1002/humu.22981
- Li
T, Miller CH, Payne AB, Craig Hooper W. The CDC Hemophilia B mutation
project mutation list: A new online resource. Mol Genet Genomic Med
2013;1(4):238-45. 10.1002/mgg3.30 https://doi.org/10.1002/mgg3.30
- Flanagan
SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict
loss-of-function and gain-of-function mutations. Genet Test Mol
Biomarkers 2010;14(4):533-7. 10.1089/gtmb.2010.0036 https://doi.org/10.1089/gtmb.2010.0036
- Kumar
P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous
variants on protein function using the SIFT algorithm. Nat Protocols
2009;4(8):1073-81. https://doi.org/10.1038/nprot.2009.86 PMid:19561590
- Choi
Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional
effect of amino acid substitutions and indels. PLoS One
2012;7(10):e46688. 10.1371/journal.pone.0046688 https://doi.org/10.1371/journal.pone.0046688
- Schwarz
JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation
prediction for the deep-sequencing age. Nat Meth 2014;11(4):361-2.
10.1038/nmeth.2890 https://doi.org/10.1038/nmeth.2890
- Shen
BW, Spiegel PC, Chang C-H, Huh J-W, Lee J-S, Kim J, Kim Y-H, Stoddard
BL. The tertiary structure and domain organization of coagulation
factor VIII. Blood 2008;111(3):1240-7. 10.1182/blood-2007-08-109918 https://doi.org/10.1182/blood-2007-08-109918
- Zögg
T, Brandstetter H. Structural Basis of the Cofactor- and
Substrate-Assisted Activation of Human Coagulation Factor IXa.
Structure 2009;17(12):1669-78. 10.1016/j.str.2009.10.011 https://doi.org/10.1016/j.str.2009.10.011
- Pinto
P, Ghosh K, Shetty S. F8 gene mutation profile in Indian hemophilia A
patients: Identification of 23 novel mutations and factor VIII
inhibitor risk association. Mutat Res 2016;786:27-33. http://dx.doi.org/10.1016/j.mrfmmm.2016.02.002 https://doi.org/10.1016/j.mrfmmm.2016.02.002
- Xue
F, Zhang L, Sui T, Ge J, Gu D, Du W, Zhao H, Yang R. Factor VIII gene
mutations profile in 148 Chinese hemophilia A subjects. Eur J Haematol
2010;85(3):264-72. 10.1111/j.1600-0609.2010.01481.x https://doi.org/10.1111/j.1600-0609.2010.01481.x
- Ahmed
R, Ivaskevicius V, Kannan M, Seifried E, Oldenburg J, Saxena R.
Identification of 32 novel mutations in the factor VIII gene in Indian
patients with hemophilia A. Haematologica 2005;90(2):283-4.
PMid:15710596
- Shekari
Khaniani M, Ebrahimi A, Daraei S, Derakhshan SM. Genotyping of Intron
Inversions and Point Mutations in Exon 14 of the FVIII Gene in Iranian
Azeri Turkish Families with Hemophilia A. Indian J Hematol Blood
Transfus 2016;32(4):475-80. 10.1007/s12288-016-0699-2 https://doi.org/10.1007/s12288-016-0699-2
- Mousavi
SH, Mesbah‐Namin SA, Rezaie N, Zeinali S. Frequencies of intron 1 and
22 inversions of factor VIII gene: A first report in Afghan patients
with severe haemophilia A. Haemophilia 2018;0(0). doi:10.1111/hae.13491
https://doi.org/10.1111/hae.13491
- Citron
M, Godmilow L, Ganguly T, Ganguly A. High throughput mutation screening
of the factor VIII gene (F8C) in hemophilia A: 37 novel mutations and
genotype–phenotype correlation. Hum Mutat 2002;20(4):267-74.
10.1002/humu.10119 https://doi.org/10.1002/humu.10119
- Guo
Z, Yang L, Qin X, Liu X, Zhang Y. Spectrum of Molecular Defects in 216
Chinese Families With Hemophilia A: Identification of Noninversion
Mutation Hot Spots and 42 Novel Mutations. Clin Appl Thromb Hemost
2017;24(1):70-8. 10.1177/1076029616687848 https://doi.org/10.1177/1076029616687848
- Lyu
C, Xue F, Liu X, Liu W, Fu R, Sun T, Wu R, Zhang L, Li H, Zhang D, Yang
R, Zhang L. Identification of mutations in the F8 and F9 gene in
families with haemophilia using targeted high-throughput sequencing.
Haemophilia 2016;22(5):e427-e34. 10.1111/hae.12924 https://doi.org/10.1111/hae.12924
- Pezeshkpoor
B, Zimmer N, Marquardt N, Nanda I, Haaf T, Budde U, Oldenburg J,
El-Maarri O. Deep intronic 'mutations' cause hemophilia A: Application
of next generation sequencing in patients without detectable mutation
in F8 cDNA. J Thromb Haemost 2013;11(9):1679-87. 10.1111/jth.12339 https://doi.org/10.1111/jth.12339
- RepessÉ
Y, Slaoui M, Ferrandiz D, Gautier P, Costa C, Costa JM, Lavergne JM,
Borel-Derlon A. Factor VIII (FVIII) gene mutations in 120 patients with
hemophilia A: Detection of 26 novel mutations and correlation with
FVIII inhibitor development. J Thromb Haemost 2007;5(7):1469-76.
10.1111/j.1538-7836.2007.02591.x https://doi.org/10.1111/j.1538-7836.2007.02591.x
- Oldenburg
J, El-Maarri O, Schwaab R. Inhibitor development in correlation to
factor VIII genotypes. Haemophilia 2002;8:23-9.
10.1046/j.1351-8216.2001.00134.x https://doi.org/10.1046/j.1351-8216.2001.00134.x
- Miller
CH, Benson J, Ellingsen D, Driggers J, Payne A, Kelly FM, Soucie JM,
Craig Hooper W, The Hemophilia Inhibitor Research Study Investigators.
F8 and F9 mutations in US haemophilia patients: Correlation with
history of inhibitor and race/ethnicity. Haemophilia 2012;18(3):375-82.
10.1111/j.1365-2516.2011.02700.x https://doi.org/10.1111/j.1365-2516.2011.02700.x
- Prescott
R, Nakai H, Saenko EL, Scharrer I, Nilsson IM, Humphries JE, Hurst D,
Bray G, Scandella D. the inhibitor antibody response is more complex in
Hemophilia A patients than in most nonhemophiliacs with factor VIII
autoantibodies. Blood 1997;89(10):3663-71. PMid:9160671
- Pipe SW. Functional roles of the factor VIII B domain. Haemophilia 2009;15(6):1187-96. 10.1111/j.1365-2516.2009.02026.x https://doi.org/10.1111/j.1365-2516.2009.02026.x
- Burke
RL, Pachl C, Quiroga M, Rosenberg S, Haigwood N, Nordfang O, Ezban M.
The functional domains of coagulation factor VIII:C. J Biol Chem
1986;261(27):12574-8. PMid:3017981
- Pittman
D, Alderman E, Tomkinson K, Wang J, Giles A, Kaufman R. Biochemical,
immunological, and in vivo functional characterization of
B-domain-deleted factor VIII. Blood 1993;81(11):2925-35. PMid:8499631
- Shelley
N, Miao-Liang L, R. TA. Some factor VIII exon 14 frameshift mutations
cause moderately severe haemophilia A. Br J Haematol
2001;115(4):977-82. doi:10.1046/j.1365-2141.2001.03173.x https://doi.org/10.1046/j.1365-2141.2001.03173.x
- Oldenburg
J, Ananyeva NM, Saenko EL. Molecular basis of haemophilia A.
Haemophilia 2004;10:133-9. 10.1111/j.1365-2516.2004.01005.x https://doi.org/10.1111/j.1365-2516.2004.01005.x
- Ogata
K, Selvaraj SR, Miao HZ, Pipe SW. Most factor VIII B domain missense
mutations are unlikely to be causative mutations for severe Hemophilia
A: Implications for genotyping. J Thromb Haemost 2011;9(6):1183-90.
10.1111/j.1538-7836.2011.04268.x https://doi.org/10.1111/j.1538-7836.2011.04268.x
- Jacquemin
M, Lavend'homme R, Benhida A, Vanzieleghem B, d'Oiron R, Lavergne J-M,
Brackmann HH, Schwaab R, VandenDriessche T, Chuah MKL, Hoylaerts M,
Gilles JGG, Peerlinck K, Vermylen J, Saint-Remy J-MR. A novel cause of
mild/moderate hemophilia A: Mutations scattered in the factor VIII C1
domain reduce factor VIII binding to von Willebrand factor. Blood
2000;96(3):958-65. PMid:10910910
- Liu
Z, Lin L, Yuan C, Nicolaes GAF, Chen L, Meehan EJ, Furie B, Furie B,
Huang M. Trp(2313)-His(2315) of factor VIII C2 domain is involved in
membrane binding: Structure of a complex between the C2 domain and an
inhibitor of membrane binding. J Biol Chem 2010;285(12):8824-9.
10.1074/jbc.M109.080168 https://doi.org/10.1074/jbc.M109.080168
- Foster
P, Fulcher C, Houghten R, Zimmerman T. Synthetic factor VIII peptides
with amino acid sequences contained within the C2 domain of factor VIII
inhibit factor VIII binding to phosphatidylserine. Blood
1990;75(10):1999-2004. PMid:2110840
- Liu
J, Zhang Y, Wang H, Huang W, Cao W, Wang X, Qu B, Wang H, Shao H, Wang
Z, Chen L, Huang W. [Molecular characterization of genetic defects in
hemophilia in Shanghai]. Zhonghua Xue Ye Xue Za Zhi 1997;18(9):464-7.
PMid:15625837
- Fidanci
ID, Kavakli K, Uçar C, Timur Ç, Meral A, Klnç Y, Saylan H, Kazanc E,
Çaglayan SH. Factor 8 (F8) gene mutation profile of Turkish hemophilia
A patients with inhibitors. Blood Coagul Fibrinolysis 2008;19(5):383-8.
10.1097/MBC.0b013e3282f9b193 https://doi.org/10.1097/MBC.0b013e3282f9b193
- Pipe
SW, Eickhorst AN, McKinley SH, Saenko EL, Kaufman RJ. Mild Hemophilia A
caused by increased rate of factor VIII A2 subunit dissociation:
Evidence for nonproteolytic inactivation of factor VIIIa in vivo. Blood
1999;93(1):176-83. PMid:9864159
- Celie
PHN, Van Stempvoort G, Jorieux S, Mazurier C, Van Mourik JA, Mertens K.
Substitution of Arg527 and Arg531 in factor VIII associated with mild
haemophilia A: Characterization in terms of subunit interaction and
cofactor function. Br J Haematol 1999;106(3):792-800.
10.1046/j.1365-2141.1999.01590.x https://doi.org/10.1046/j.1365-2141.1999.01590.x
- Pieters
J, Lindhout T, Hemker H. In situ-generated thrombin is the only enzyme
that effectively activates factor VIII and factor V in
thromboplastin-activated plasma. Blood 1989;74(3):1021-4. PMid:2502206
- Nogami
K, Zhou Q, Wakabayashi H, Fay PJ. Thrombin-catalyzed activation of
factor VIII with His substituted for Arg372 at the P(1) site. Blood
2005;105(11):4362-8. 10.1182/blood-2004-10-3939 https://doi.org/10.1182/blood-2004-10-3939
- Shima
M, Ware J, Yoshioka A, Fukui H, Fulcher C. An arginine to cysteine
amino acid substitution at a critical thrombin cleavage site in a
dysfunctional factor VIII molecule. Blood 1989;74(5):1612-7.
PMid:2506948
- Pittman
DD, Kaufman RJ. Proteolytic requirements for thrombin activation of
anti-hemophilic factor (factor VIII). Proc Natl Acad Sci U S A
1988;85(8):2429-33. https://doi.org/10.1073/pnas.85.8.2429
- Ma
GC, Chang SP, Chen M, Kuo SJ, Chang CS, Shen MC. The spectrum of the
factor 8 (F8) defects in Taiwanese patients with haemophilia A.
Haemophilia 2008;14(4):787-95. 10.1111/j.1365-2516.2008.01687.x https://doi.org/10.1111/j.1365-2516.2008.01687.x
- Radic
CP, Rossetti LC, Abelleyro MM, Candela M, Bianco RP, Pinto MdT, Larripa
IB, Goodeve A, De Brasi CD. Assessment of the F9 genotype-specific FIX
inhibitor risks and characterization of 10 novel severe F9 defects in
the first molecular series of Argentine patients with haemophilia B.
Thromb Haemost 2013;109(1):24-33. 10.1160/TH12-05-0302 https://doi.org/10.1160/TH12-05-0302
- Baralle
D, Baralle M. Splicing in action: Assessing disease causing sequence
changes. J Med Genet 2005;42(10):737-48. 10.1136/jmg.2004.029538 https://doi.org/10.1136/jmg.2004.029538
- Hamasaki-Katagiri
N, Salari R, Simhadri VL, Tseng SC, Needlman E, Edwards NC, Sauna ZE,
Grigoryan V, Komar AA, Przytycka TM, Kimchi-Sarfaty C. Analysis of F9
point mutations and their correlation to severity of haemophilia B
disease. Haemophilia 2012;18(6):933-40.
10.1111/j.1365-2516.2012.02848.x https://doi.org/10.1111/j.1365-2516.2012.02848.x
- Wulff
K, Bykowska K, Lopaciuk S, Herrmann FH. Molecular analysis of
hemophilia B in Poland: 12 novel mutations of the factor IX gene. Acta
Biochim Pol 1999;46(3):721-6. PMid:10698280
- Li
X, Drost JB, Roberts S, Kasper C, Sommer SS. Factor IX mutations in
South Africans and African Americans are compatible with primarily
endogenous influences upon recent germline mutations. Hum Mutat
2000;16(4):371-. https://doi.org/10.1002/1098-1004(200010)16:4<371::AID-HUMU11>3.0.CO;2-P
- Yu
T, Dai J, Liu H, Ding Q, Lu Y, Wang H, W ang X, Fu Q. Spectrum of F9
mutations in Chinese haemophilia B patients: Identification of 20 novel
mutations. Pathology 2012;44(4):342-7. 10.1097/PAT.0b013e328353443d https://doi.org/10.1097/PAT.0b013e328353443d
- Di Scipio RG, Kurachi K, Davie EW. Activation of human factor IX (Christmas factor). J Clin Invest 1978;61(6):1528-38. https://doi.org/10.1172/JCI109073 PMid:659613 PMCid:PMC372679
- Attali
O, Vinciguerra C, Trzeciak MC, Durin A, Pernod G, Gay V, Ménart C,
Sobas F, Dechavanne M, Négrier C. Factor IX gene analysis in 70
unrelated patients with Haemophilia B: Description of 13 new mutations.
Thromb Haemost 1999;82(5):1437-42. PMid:10595634
- Jayandharan
GR, Shaji RV, Baidya S, Nair SC, Chandy M, Srivastava A. Molecular
characterization of factor IX gene mutations in 53 patients with
haemophilia B in India. Thromb Haemost 2005;94(4):883-6. PMid:16270648
[TOP]