Mario Sabatelli1,4, Luca Laurenti2,4 and Marco Luigetti3,4.
1 Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italia. Centro Clinico NEMO Adulti.
2 Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italia. UOC Ematologia.
3 Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italia. UOC Neurologia.
4 Università Cattolica del Sacro Cuore, Roma, Italia.
Correspondence to: Dr Mario Sabatelli. Centro Clinico NEMO Adulti,
Roma; Institute of Neurology, Catholic University of the Sacred Heart,
Rome, Italy Largo A Gemelli 8, 00168 ROME – ITALY. Tel.:
+39-06-30158220 Fax No. : +39-06-30158266. E-mail:
mario.sabatelli@unicatt.it
Published: September 1, 2018
Received: June 27, 2018
Accepted: August 6, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018057 DOI
10.4084/MJHID.2018.057
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Peripheral
neuropathies are a vast group of diseases with heterogeneous
aetiologies, including genetic and acquired causes. Several
haematological disorders may cause an impairment of the peripheral
nervous system, with diverse mechanisms and variable clinical,
electrophysiological and pathological manifestations. In this practical
review, we considered the main phenotypes of peripheral nervous system
diseases associated with lymphoproliferative disorders.
The area
of intersection of neurological and haematological fields is of
particular complexity and raises specific problems in the clinical
practice of lymphoproliferative disorders. The personal crosstalk
between neurologists and haematologists remains a fundamental tool for
a proper diagnostic process which may lead to successful treatments in
most cases.
|
Introduction
The
peripheral nervous system (PNS) consists of sensory, motor, and
autonomic neurons that lie outside the confines of the central nervous
system. It is a conceptual artifice rather than a concrete anatomical
definition, as most neurons with peripheral projection lie partly
within the peripheral and partly within the central nervous system.
Peripheral
neuropathies are diseases with heterogeneous aetiologies, including
genetic and acquired causes. Several haematological disorders may cause
an impairment of the peripheral nervous system, with diverse mechanisms
and variable clinical, electrophysiological and pathological
manifestations.
Peripheral neuropathies may be classified into
distinct phenotypes based on different clinical, electrophysiological
and pathological criteria. Clinically, we distinguish two great groups:
symmetric polyneuropathies and focal neuropathies. Both groups may be
further differentiated in demyelinating and axonal forms, according to
the predominant target of the disease process that is myelin sheath or
axon, respectively.
In this practical review, we considered the
main phenotypes of peripheral nervous system diseases and the
lymphoproliferative disorders which may be associated with.
Focal Neuropathies
In
several acquired and hereditary conditions, the involvement of the PNS
may be limited to a single nerve (mononeuropathy) orto a few nerves
(multiple mononeuropathy or multifocal neuropathy). This condition can
be easily differentiated from symmetric polyneuropathy on the clinical
ground, but the electrophysiological examination is helpful in
confirming the multifocal nature of nerve involvement and may indicate
the presence of predominant or exclusive axonal or demyelinating
pattern.
Vasculitic neuropathy.
Primary and secondary vasculitis are a leading cause of focal
neuropathies and should firstly be considered in the differential
diagnosis of focal neuropathies, especially if painful. In a minority
of cases vasculitis give rise to a generalized, but asymmetric,
neuropathy.[1] Inflammation of the small epineural
arteries leads to their occlusion with a secondary ischemic injury of
the nerve fibres. Vasculitic neuropathies may occur in the context of
widespread, multiorgan involvement, or, less commonly, may represent
the only manifestation of the disease (non-systemic vasculitic
neuropathies). Haematological diseases associated with vasculitis
neuropathy include cryoglobulinemia, eosinophilic granulomatosis with
polyangiitis (formerly named Churg-Strauss disease), and some forms of
paraproteinemias (Figure 1).
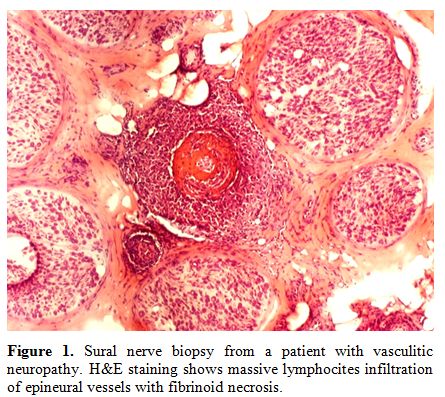 |
Figure 1. Sural nerve
biopsy from a patient with primary multifocal lymphoma of peripheral
nervous system. Immunohistochemistry with anti-CD20 (green) and DAPI
(blue) confirms diffuse infiltration of one nerve fascicle by
lymphomatous cells. |
Cryoglobulinemia
refers to the presence of circulating immunoglobulins that precipitate
at cold temperatures. Types II mixed cryoglobulinemia is the form most
frequently associated with neuropathy. Hepatitis C virus (HCV) is
present in 80–90% of patients with types II of cryoglobulinemia. When
no underlying disorder is detected, the condition is termed essential.
In patients with HCV-related cryoglobulinemia, about 65% develop a
neuropathy.[2] However, most cases have a distal,
predominantly sensory polyneuropathy with a very slowly progressive
course, which is related to HCV infection per se
rather than to vasculitic damage. Additional features of the
cryoglobulinemic syndrome include purpura, skin ulcers, arthralgias,
sicca syndrome, Raynaud’s phenomenon, glomerulonephritis, and
lymphadenopathy. Rheumatoid factors and hypocomplementemia occur in the
majority of patients.
Churg-Strauss disease is characterized by
blood eosinophilia greater than 10%, asthma, pulmonary infiltrates.
Involvement of PNS occurs in 60-70% of patients.[3]
Finally, vasculitis may be one of the mechanisms by which IgM paraproteinemia damages the peripheral nerves (see after).[4-5]
Vasculitis should be considered when acute, focal nerve lesion occurs
in the setting of the classic indolent IgM polyneuropathy or in
asymptomatic individuals.
Neurolymphomatosis.
Infiltration by lymphomatous cells of the PNS is a rare and frequently
ignored complication of non-Hodgkin lymphoma. Direct invasion of
lymphoma cells into the PNS may occur in patients with a previous
diagnosis of lymphoma but may represent the first and unique
manifestation of the haematological malignancy, a condition defined as
primary neurolymphomatosis.
Nerve roots and plexi are more frequently involved; other sites include
cranial nerves, sciatic nerve and cauda equine. Lymphomatous cell
invasion induces demyelination and subsequent axonal degeneration in
the portion distal from the infiltration site. Differential diagnosis
with inflammatory radiculo-plexo-neuropathies and other forms of focal
inflammatory neuropathies is challenging. Severe pain and the
progressive course despite immunomodulating
therapies should raise the suspicion. Total-body
fluorine-18 fluorodeoxyglucose (18F-FDG) positron emission
tomography-computed tomography (PET-CT) is sensitive though not
specific imaging technique. Targeted fascicular nerve biopsy is the
only tool able to provide a definitive diagnosis (Figure 2).[6-7]
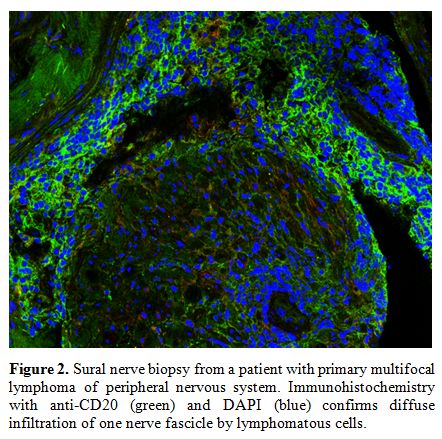 |
Figure 2. Sural nerve
biopsy from a patient with primary multifocal lymphoma of peripheral
nervous system. Immunohistochemistry with anti-CD20 (green) and DAPI
(blue) confirms diffuse infiltration of one nerve fascicle by
lymphomatous cells. |
Immunoglobulin infiltration.
Multiple mononeuritis have been described as the predominant clinical
manifestation in rare patients with Waldenström's macroglobulinaemia in
which the underlying mechanism is a massive light and heavy chain
deposition within the nerves resulting in massive fascicular hyalinosis
(Figure 3 A-D).[4,8-9]
In these cases, protein accumulation in the endoneurium and epineurium
behaves differently from amyloid as it does not stain with Congo Red.[4,8-9]
The presence of polyneuropathy is currently considered an indication to
start treatment in smouldering Waldenström macroglobulinemia.[10]
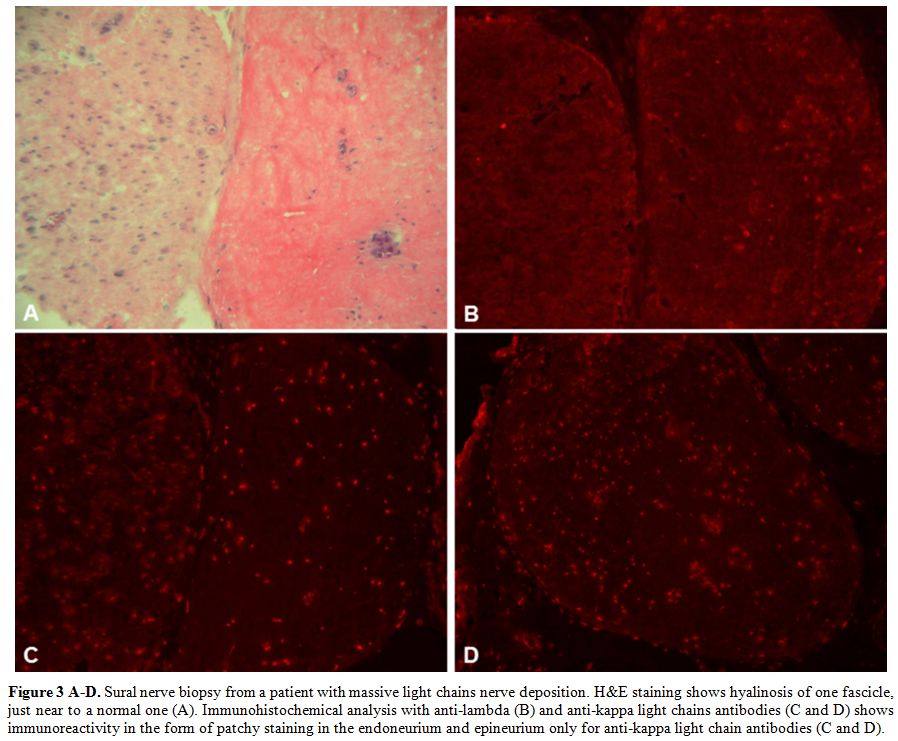 |
Figure 3 A-D. Sural nerve
biopsy from a patient with massive light chains nerve deposition.
H&E staining shows hyalinosis of one fascicle, just near to a
normal one (A). Immunohistochemical analysis with anti-lambda (B) and
anti-kappa light chains antibodies (C and D) shows immunoreactivity in
the form of patchy staining in the endoneurium and epineurium only for
anti-kappa light chain antibodies (C and D). |
Others.
Focal neuropathies due to direct infiltration of malignant cells have
been occasionally reported in patients with acute myeloid leukaemia.[11] Inflammatory asymmetric radiculoplexopathy is a very rare complication of stem cell transplantation.[12]
Demyelinating and Axonal Polyneuropathies
Peripheral
nerve disorders are traditionally classified as primary demyelinating
or axonal. In demyelinating neuropathies, segments of myelin sheath may
be damaged leaving the axon anatomically intact. On the contrary, in
axonal neuropathies, there is a loss of motor or sensory axons while
the myelin sheath of the surviving fibres is normal. The
electrophysiological examination is able to differentiate demyelinating
from axonal neuropathies,[13] thus helping in clinical practice and offering clues since causes of demyelinating and of axonal forms are different.
Demyelinating polyneuropathies.
The myelin sheath is the primary target in numerous genetic and
acquired conditions, the latter including Chronic Inflammatory
Demyelinating Polyradiculoneuropathy (CIDP), Demyelinating
Polyneuropathy associated with IgM paraproteinemia, POEMS
(polyneuropathy, organomegaly, endocrinopathy, M protein, and skin
changes) syndrome, and Guillain Barré Syndrome (GBS).
Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Guillain Barré Syndrome.
CIDP and GBS are immune-mediated diseases of peripheral nerves, usually
occurring as isolated conditions. There is evidence that they may be
associated with haematological diseases, particularly lymphoma, which
may act as predisposing conditions.
CIDP may have a chronically progressive or relapsing course and responds to immune modulating or immunosuppressive treatments.[14]
In typical CIDP there is a symmetric motor/sensory disorder with
proximal and distal weakness and areflexia with electrophysiological
signs of demyelination, including conduction slowing, temporal
dispersion and/or conduction block. Although abnormalities in both
cellular and humoral immunity have been shown, the causes of CIDP
remain largely unknown, and no specific antigen has been identified.
Recently, antibodies against paranodal axo-glial proteins, resulting in
conduction alteration without overt demyelination, have been detected
in demyelinating neuropathies, but these conditions represent a
distinct subgroup of CIDP. Near always CIDP is an idiopathic condition,
while in a minority of cases it has been described in association with
various disorders and in these cases, it may be more difficult to
recognise. A recent review has shown that haematological diseases,
particularly non-Hodgkin lymphoma, are the malignancies most commonly
associated with CIDP. CIDP frequently precedes the haematological
diagnosis and responds to treatments in the same manner as the
idiopathic form.[15]
PNS involvement occurs in
about 5-14% of patients with lymphoma. Besides the nerve infiltration
and CIDP, mentioned above, other causes are chemotherapy-induced
neuropathy, amyloid neuropathy, paraneoplastic neuropathy and
Varicella-Zoster (VZV) infection of ganglia/nerves.[16-17]
Since CIDP is a condition responding to specific treatments, a careful
electrophysiological examination is required to identify the presence
of demyelinating features typical of CIDP.
Guillain-Barre syndrome
(GBS) is an acute disorder affecting peripheral nerves and nerve roots
with maximum severity within four weeks from disease onset. GBS may be
classified into variants based on clinical features and
electrodiagnostic findings. The most common variant is acute
inflammatory demyelinating polyradiculoneuropathy (AIDP), while acute
motor axonal neuropathy (AMAN), acute motor and sensory axonal
neuropathy (AMSAN), Miller Fisher syndrome,
pharyngeal-cervical-brachial variant, cranial polyneuritis, and acute
pandysautonomia account for a minority of cases.
GBS is usually an idiopathic condition but in rare cases is associated with lymphoma[16] and in even much rare cases precedes the haematological disease.[18]
GBS
has rarely described after stem cell transplantation for haematological
diseases, possibly caused by an immune reconstitution syndrome or by a
paraneoplastic phenomenon.
Finally, in rare cases, an acute
polyradiculoneuropathy mimicking GBS may be the presenting
manifestation of acute myeloid leukaemia.[19,20]
Chronic Demyelinating symmetric polyneuropathy and paraproteinemia.
Coexistence of neuropathy and monoclonal gammopathy represents a common
and challenging problem in clinical practice. Monoclonal proteins may
occur in the context of several haematologic malignancies or, more
frequently, as a monoclonal gammopathy of undetermined significance
(MGUS). MGUS is relatively common in the general population, with a
prevalence of 3% to 4% among individuals older than 50 years,[21]
so that its presence in a patient with neuropathy may be coincidental.
Furthermore, 11% of individuals with MGUS show a progression to
multiple myeloma or another plasma-cell or lymphoid disorder over the
time.[22] Finally, the relationship between
paraproteinemia and neuropathy is multifaceted, as several pathogenic
mechanisms are involved with variable clinical, electrophysiological
pathologic manifestations. Depending on the type of M protein three
major types of MGUS are considered: IgM MGUS, non-IgM MGUS (which
includes IgG and IgA MGUS), and light-chain MGUS.
If the
paraproteinemia is detected in association with a demyelinating
polyneuropathy, diagnostic and pathogenic consideration differ for each
of these three groups:
a) IgM MGUS and demyelinating neuropathy:
- Demyelinating neuropathy and IgM paraproteinemia.
IgM paraproteinemic neuropathy represents an established and
well-characterised entity, as it shows peculiar clinical,
electrophysiological and pathologic characteristics, which allow a
clear differentiation from CIDP.[23-24] The clinical
picture in that of “distal acquired demyelinating symmetric” (DADS)
sensory and motor neuropathy. Most patients have distal paresthesias,
sensory ataxia, frequent hand tremor with little or no weakness of
tibia-peroneal muscles. The neuropathy has an indolent course, usually
with little functional impairment over time. If in this context a rapid
deterioration occurs, the possibility of a vasculitic injury should be
considered. Electrophysiological signs are also characteristic and
include slowing of conduction velocities with particularly increased
distal motor latencies, without conduction blocks. Pathologic
examination of nerve biopsy shows typical widening of myelin lamellae
and irregular myelin folding while little or no demyelination is
observed (Figure 4 A-B). In about 50% of patients with IgM paraproteinemic neuropathies, the M protein binds to myelin-associated glycoprotein (MAG).[23-24] In anti-MAG negative patients[25-26] reactivity against gangliosides and their complexes were detected in 35% of cases.
This
neuropathy does not respond to many of the immune therapies that are
effective in CIDP. There is very low-quality evidence of benefit from
rituximab.[27]
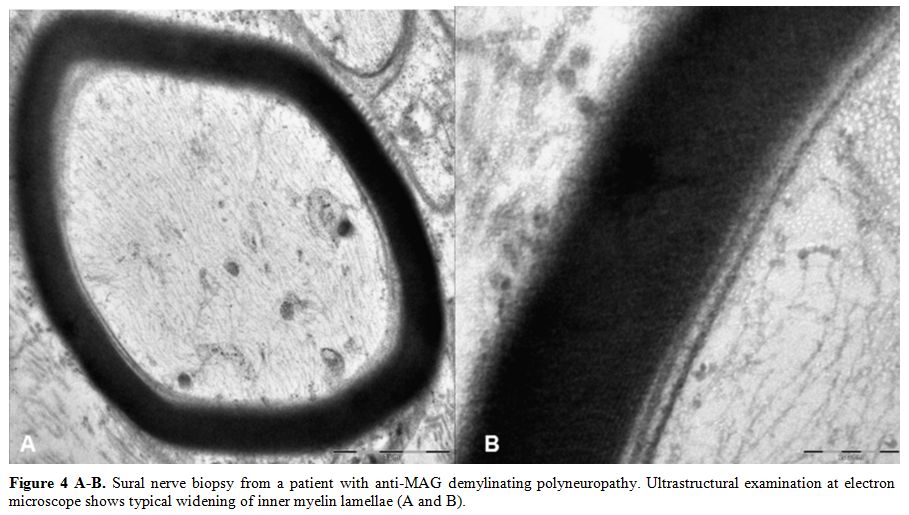 |
Figure 4 A-B. Sural nerve
biopsy from a patient with anti-MAG demylinating polyneuropathy.
Ultrastructural examination at electron microscope shows typical
widening of inner myelin lamellae (A and B). |
- Demyelinating neuropathy with IgM paraproteinemia and Ophthalmoplegia.
Chronic Ataxic Neuropathy with Ophthalmoplegia, M-protein, cold
Agglutinins and Disialosyl antibodies (anti-ganglioside, anti-GD1b, and
anti-GQ1b) (CANOMAD syndrome) is a rare phenotype associated with an
IgM MGUS.[28] The clinical picture is characterised
by a chronic neuropathy with marked sensory ataxia and areflexia, and
with relatively preserved motor function in the limbs. In addition, 90%
of cases have oculomotor and bulbar muscles impairment as fixed or as
relapsing-remitting features. The IgM antibodies are cold agglutinins
in 50% of cases.
b) Non-IgM MGUS and demyelinating neuropathy:
- Demyelinating neuropathy with IgG or IgA paraproteinemia.
When a non-IgM paraproteinemia is found in a context of a demyelinating
neuropathy, the most likely situation is that it represents a casual
combination of an otherwise typical CIDP with an MGUS. This explanation
is based on the observation that the neuropathy has no peculiar
clinical, electrophysiological or pathological aspect and no reactivity
to nerve antigen has been observed in these cases. Responses to therapy
are not different in CIDP patients with or without IgG or IgA
paraproteinemia.[29]
- Demyelinating neuropathy with IgG or IgA paraproteinemia, with atypical aspects.
When a demyelinating neuropathy associated with non-IgM monoclonal
component behave differently from typical CIDP, the POEMS syndrome
should be considered. In this condition, a multitude of clinical and
laboratory signs may accompany the polyneuropathy. Clinical alerts
include skin changes (hyperpigmentation, hypertrichosis, acrocyanosis,
flushing, white nails), oedema, pleural effusion or ascites,
papilledema, weight loss, severe muscular deterioration not responding
to usual therapies used in CIDP. The detection of
thrombocytosis/polycythaemia and/or organomegaly (splenomegaly,
hepatomegaly or lymphadenopathy) are additional elements. The
electrophysiological examination also shows a more severe pattern than
in CIDP, with marked reduction of motor amplitudes and presence of
fibrillation potentials, expression of severe axonal loss. The M
protein is IgG or IgA, almost always λ. The evidence of osteosclerotic
bone lesions at radiologic skeletal studies and of elevation of
vascular endothelial growth factor (VEGF) are crucial for the
diagnosis. The diagnosis of POEMS syndrome is made in a demyelinating
polyneuropathy with M protein when one of the major criteria and at
least one of the minor criteria are present.[30] In conclusion, though POEMS is easily mistaken for CIDP, the diagnosis is relatively simple if it is considered.
c) Light-chain MGUS and demyelinating neuropathies:
AL
amyloidosis is an axonal neuropathy (see after), but in some cases, a
slight slowing of conduction velocities and an increase of distal and F
waves latencies may be misleading, and CIDP may be erroneously
diagnosed.[31]
Axonal neuropathies.
Axonal neuropathies encompass a vast group of genetic and acquired
conditions, whose etiologic definition is more difficult than for
demyelinating neuropathies. Three main groups of axonal neuropathies
may be present in haematological disorders: sensory neuronopathies,
length-dependent polyneuropathies, and axonal polyneuropathy associated
with paraproteinemia.
Sensory neuronopathies.
Sensory neuronopathies (SNs) or ganglionopathies encompass a group of
disorders characterised by primary degeneration of sensory neurons
whose cell body is located in the dorsal root ganglia (DRG). These
cells are particularly susceptible to circulating agents, including
antibodies or toxins, because capillaries are fenestrated in the DRG,
and the blood-nerve barrier is looser than normally. On the contrary,
in nerve trunks, capillaries are not fenestrated, and endothelial cells
are united by tight junctions resulting in a true blood-nerve barrier
with a restricted permeability. SNs represent the most frequent
manifestation of paraneoplastic neurological syndromes,[32]
but may also be caused by immunologic diseases as Sjögren syndrome, by
HIV, EBV, VZV, HTLV-1 virus infection and by toxic agents as
pyridoxine, cisplatin, carboplatin, oxaliplatin.[33]
The typical clinical manifestation is an asymmetric,
non-length-dependent sensory impairment with subacute onset. Sensory
ataxic or painful neuropathy may be predominant, depending on the type
of neurons involved. Chronic forms also exist and are generally
idiopathic. Paraneoplastic sensory neuronopathy is associated with
small-cell lung cancer and less frequently with bronchial carcinoma,
breast and ovarian cancer. HL and NHLs may rarely cause paraneoplastic
neurological syndromes, mainly cerebellar degeneration and
dermato/polymyositis while sensory neuronopathy is reported in very few
cases. Notably, onconeural antibodies which are detected in the
majority of cases of paraneoplastic neuropathies associated with small
cell lung carcinoma are generally absent in patients with lymphoma.[34,35]
Length-dependent axonal polyneuropathy.
In chronic axonal neuropathies, cell bodies of neurons remain intact,
at least in the initial phases of the disease, but the axons are
impaired in proportion to their length. Since the longest axons are
affected earlier, motor and sensory signs begin symmetrically in distal
legs and progress to involve distal regions of upper limbs and
eventually proximal regions. Electrophysiological examination shows a
reduction of the amplitude of sensory and motor potentials while the
conduction time (conduction velocities, distal and F wave latencies) is
average or slightly altered.
A major problem in clinical practice
is that in a significant proportion of patients with chronic axonal
neuropathy no aetiology can be identified, despite extensive
investigations. This condition, termed Chronic Idiopathic Axonal
Polyneuropathy (CIAP), affects people usually in the sixth-seventh
decades of life and accounts for about 30% of neuropathy cases.[36]
CIAP is characterised by prominent or isolated sensory symptoms with
numbness and tingling in the feet, or sensory ataxia, while motor
impairment is less severe.[37] CIAP has an indolent and usually “benign” course with phases of stabilisation.
Axonal
sensory-motor polyneuropathy is a rare manifestation of a
paraneoplastic syndrome and the association with lymphoma has been
described in isolated cases.[34]
Axonal polyneuropathy associated with paraproteinemia.
Since CIAP and MGUS are highly prevalent in people over 50 years of
age, the possibility of a chance association should be considered.
Accurate follow-up is recommended in order to detect possible changes
in both conditions.
If the polyneuropathy shows a course which
appears atypical for CIAP and in particular if motor weakness and
atrophy occur in legs and hands, or painful paraesthesias become
prominent, the possibility of AL amyloidosis must be considered. Other
critical red flags include weight loss, renal involvement, diarrhoea
alternated with constipation, postural hypotension, cardiomyopathy. The
diagnosis of amyloidosis is straightforward if considered.
Primary
systemic AL amyloidosis is a disease characterised by diffuse
deposition of amyloid fibrils derived from immunoglobulin light chains
which, instead of forming the α-helical
configuration, became misfolded and forms a β-pleated sheet. Amyloid
deposits are found mainly in the heart, kidney, gastrointestinal tract,
and peripheral nervous system. AL amyloidosis may be secondary to
Multiple Myeloma in 10% of cases. Twenty per cent of patients with
systemic AL amyloidosis present with an axonal sensory-motor and
autonomic polyneuropathy.[30] The monoclonal protein is usually IgG or IgA, but in a minority of cases, AL amyloidosis is associated with IgM.[38-39]
Diagnosis
is based on the demonstration of amyloid deposits, confirmed by Congo
Red staining, in heart, peripheral nerves, rectum, abdominal fat or
salivary glands (Figure 5 A-B).
Importantly, the presence of an MGUS in patients with systemic
amyloidosis does not necessarily imply a diagnosis of primary AL
amyloidosis, as hereditary amyloidosis may be identified in about 10%
per cent of patients with a presumptive diagnosis of AL amyloidosis.[40-41]
Late-onset, sporadic, familial amyloid polyneuropathy caused by TTR
(transthyretin) gene mutations may be easily misdiagnosed as AL,[42]
so mutational analysis of TTR should not be overlooked when managing
amyloidosis, also considering the new promising therapeutic options.[43-45]
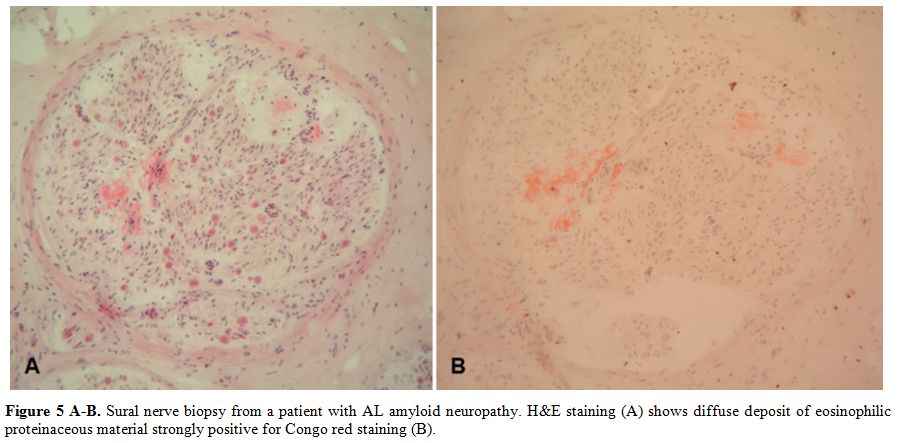 |
Figure 5 A-B. Sural nerve
biopsy from a patient with AL amyloid neuropathy. H&E staining (A)
shows diffuse deposit of eosinophilic proteinaceous material strongly
positive for Congo red staining (B). |
Conclusions
The
area of intersection of neurological and haematological fields is of
particular complexity and raises several problems in clinical practice,
mainly when the peripheral nervous system is involved, as happens with
a certain frequency in some lymphoproliferative diseases.
A proper
cultural approach is needed as in most conditions the diagnosis is
simple if considered. The personal crosstalk between neurologists and
haematologists remains a fundamental tool for a proper diagnostic
process which may lead to successful treatments in most cases.
References
- Collins MP, Dyck PJ, Gronseth GS, Guillevin L,
Hadden RD, Heuss D, Léger JM, Notermans NC, Pollard JD, Said G, Sobue
G, Vrancken AF, Kissel JT; Peripheral Nerve Society. Peripheral Nerve
Society Guideline on the classification, diagnosis, investigation, and
immunosuppressive therapy of non-systemic vasculitic neuropathy:
executive summary. J Peripher Nerv Syst. 2010;15:176-184.
 https://doi.org/10.1111/j.1529-8027.2010.00281.x PMid:21040139
https://doi.org/10.1111/j.1529-8027.2010.00281.x PMid:21040139
- Gwathmey KG, Burns TM, Collins MP, Dyck PJ. Vasculitic neuropathies. Lancet Neurol. 2014;13:67-82. https://doi.org/10.1016/S1474-4422(13)70236-9
- Comarmond
C, Pagnoux C, Khellaf M, et al. Eosinophilic granulomatosis with
polyangiitis (Churg-Strauss): clinical characteristics and long-term
followup of the 383 patients enrolled in the French Vasculitis Study
Group cohort. Arthritis Rheum 2013;65:270-281. https://doi.org/10.1002/art.37721 PMid:23044708
- Luigetti
M, Conte A, Montano N, Del Grande A, Madia F, Lo Monaco M, Laurenti L,
Sabatelli M. Clinical and pathological heterogeneity in a series of 31
patients with IgM-related neuropathy. J Neurol Sci. 2012;319:75-80 https://doi.org/10.1016/j.jns.2012.05.012 PMid:22632783
- Vital
A, Favereaux A, Martin-Dupont P, Taupin JL, Petry K, Lagueny A, Canron
MH, Vital C. Anti-myelin-associated glycoprotein antibodies and
endoneurial cryoglobulin deposits responsible for a severe neuropathy.
Acta Neuropathol. 2001;102:409-412. PMid:11603819
- Tomita
M, Koike H, Kawagashira Y, Iijima M, Adachi H, Taguchi J, Abe T, Sako
K, Tsuji Y, Nakagawa M, Kanda F, Takeda F, Sugawara M, Toyoshima I,
Asano N, Sobue G. Clinicopathological features of neuropathy associated
with lymphoma. Brain. 2013;136:2563-2578. https://doi.org/10.1093/brain/awt193 PMid:23884813
- Del
Grande A, Sabatelli M, Luigetti M, Conte A, Granata G, Rufini V, Del
Ciello A, Gaudino S, Fernandez E, Hohaus S, Coli A, Lauriola L. Primary
multifocal lymphoma of peripheral nervous system: case report and
review of the literature. Muscle Nerve. 2014;50:1016-1022. https://doi.org/10.1002/mus.24354 PMid:25088432
- Figueroa
JJ, Bosch EP, Dyck PJ, Singer W, Vrana JA, Theis JD, Dogan A, Klein CJ.
Amyloid-like IgM deposition neuropathy: a distinct clinico-pathologic
and proteomic profiled disorder. J Peripher Nerv Syst. 2012
Jun;17:182-190. https://doi.org/10.1111/j.1529-8027.2012.00406.x PMid:22734903 PMCid:PMC3895329
- Vital
C, Deminiere C, Bourgouin B, Lagueny A, David B, Loiseau P.
Waldenström's macroglobulinemia and peripheral neuropathy: deposition
of M-component and kappa light chain in the endoneurium. Neurology
1985;35:603-606. https://doi.org/10.1212/WNL.35.4.603 PMid:2984603
- Gertz MA. Waldenström macroglobulinemia treatment algorithm 2018. Blood Cancer J. 2018;8:40. https://doi.org/10.1038/s41408-018-0076-5 PMid:29712895 PMCid:PMC5928091
- Yiu
CR, Lee LH, Kumar PM, Wong GC. A patient with extramedullary acute
myeloid leukaemia involving the brachial plexus: Case report and review
of the literature. Turk J Haematol. 2008;25:98-100. PMid:27264448
- Karam
C, Mauermann ML, Johnston PB, Lahoria R, Engelstad JK, Dyck PJ.
Immune-mediated neuropathies following stem cell transplantation. J
Neurol Neurosurg Psychiatry. 2014;85:638-642. https://doi.org/10.1136/jnnp-2013-306657 PMid:24273223
- Joint
Task Force of the EFNS and the PNS. European Federation of Neurological
Societies/Peripheral Nerve Society Guideline on management of chronic
inflammatory demyelinating polyradiculoneuropathy: report of a joint
task force of the European Federation of Neurological Societies and the
Peripheral Nerve Society--First Revision. J Peripher Nerv Syst.
2010;15:1-9. https://doi.org/10.1111/j.1529-8027.2010.00245.x PMid:20433600
- Lewis RA. Chronic inflammatory demyelinating polyneuropathy. Curr Opin Neurol. 2017;30:508-512. https://doi.org/10.1097/WCO.0000000000000481 PMid:28763304
- Rajabally
YA, Attarian S. Chronic inflammatory demyelinating polyneuropathy and
malignancy: A systematic review. Muscle Nerve. 2018;57:875-883. https://doi.org/10.1002/mus.26028 PMid:29194677
- Stübgen JP. Lymphoma-associated dysimmune polyneuropathies. J Neurol Sci. 2015;355:25-36. https://doi.org/10.1016/j.jns.2015.06.003 PMid:26070654
- Hughes
RA, Britton T, Richards M. Effects of lymphoma on the peripheral
nervous system. J R Soc Med. 1994;87:526-530. PMid:7932460
PMCid:PMC1294770
- Anderson
D, Beecher G, Steve TA, Jen H, Camicioli R, Zochodne DW. Neurological
Nuance: Hodgkin lymphoma presenting with Guillain-BarrÉ syndrome.
Muscle Nerve.2017;55:601-604. https://doi.org/10.1002/mus.25439 PMid:27756115
- Hirst
CL, Willis M, Hussain H, Powell R. Acute myeloid leucaemia presenting
as a rapidly progressive polyradiculoneuropathy. BMJ Case Rep.
2015;2015.
- Kostic
I, Ruiz M, Branca A, Nabergoj M, Piazza F, Semenzato G, Gurrieri C,
Briani C. Possible neuroleukemiosis in two patients with acute myeloid
leukemia in complete bone marrow remission. J Neurol Sci. 2018 Jun
30;392:63-64. doi: 10.1016/j.jns.2018.06.029. [Epub ahead of print] https://doi.org/10.1016/j.jns.2018.06.029
- Kyle
RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR,
Dispenzieri A, Katzmann JA, Melton LJ 3rd. Prevalence of monoclonal
gammopathy of undetermined significance. N Engl J Med.
2006;354:1362-1369. https://doi.org/10.1056/NEJMoa054494 PMid:16571879
- Kyle
RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, Rajkumar
SV. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined
Significance. N Engl J Med. 2018;378:241-249. https://doi.org/10.1056/NEJMoa1709974 PMid:29342381 PMCid:PMC5852672
- Nobile-Orazio
E, Marmiroli P, Baldini L, Spagnol G, Barbieri S, Moggio M, Polli N,
Polli E, Scarlato G. Peripheral neuropathy in macroglobulinemia:
incidence and antigen-specificity of M proteins. Neurology.
1987;37:1506-1514. https://doi.org/10.1212/WNL.37.9.1506 PMid:2442666
- Nobile-Orazio E. IgM paraproteinaemic neuropathies. Curr Opin Neurol. 2004;17:599-605. https://doi.org/10.1097/00019052-200410000-00010 PMid:15367864
- Stork
AC, Jacobs BC, Tio-Gillen AP, Eurelings M, Jansen MD, van den Berg LH,
Notermans NC, van der Pol WL. Prevalence, specificity and functionality
of anti-ganglioside antibodies in neuropathy associated with IgM
monoclonal gammopathy. J Neuroimmunol. 2014;268:89-94. https://doi.org/10.1016/j.jneuroim.2014.01.012 PMid:24529728
- Stork
AC, Lunn MP, Nobile-Orazio E, Notermans NC. Treatment for IgG and IgA
paraproteinaemic neuropathy. Cochrane Database Syst Rev.
2015;(3):CD005376. https://doi.org/10.1002/14651858.CD005376.pub3
- Lunn
MP, Nobile-Orazio E. Immunotherapy for IgM anti-myelin-associated
glycoprotein paraprotein-associated peripheral neuropathies. Cochrane
Database Syst Rev. 2016;10:CD002827. https://doi.org/10.1002/14651858.CD002827.pub4
- Willison
HJ, O'Leary CP, Veitch J, Blumhardt LD, Busby M, Donaghy M, Fuhr P,
Ford H, Hahn A, Renaud S, Katifi HA, Ponsford S, Reuber M, Steck A,
Sutton I, Schady W, Thomas PK, Thompson AJ, Vallat JM, Winer J. The
clinical and laboratory features of chronic sensory ataxic neuropathy
with anti-disialosyl IgM antibodies. Brain. 2001;124:1968-1977. https://doi.org/10.1093/brain/124.10.1968 PMid:11571215
- Dyck
PJ, Low PA, Windebank AJ, Jaradeh SS, Gosselin S, Bourque P, Smith BE,
Kratz KM, Karnes JL, Evans BA, et al. Plasma exchange in polyneuropathy
associated with monoclonal gammopathy of undetermined significance. N
Engl J Med. 1991;325:1482-1486. https://doi.org/10.1056/NEJM199111213252105 PMid:1658648
- Dispenzieri A. POEMS syndrome: 2017 Update on diagnosis, risk stratification, and management. Am J Hematol. 2017;92:814-829. https://doi.org/10.1002/ajh.24802 PMid:28699668
- Luigetti
M, Papacci M, Bartoletti S, Marcaccio A, Romano A, Sabatelli M. AL
amyloid neuropathy mimicking a chronic inflammatory demyelinating
polyneuropathy. Amyloid. 2012;19:53-55. https://doi.org/10.3109/13506129.2011.650247 PMid:22292918
- Antoine JC, Camdessanché JP. Paraneoplastic neuropathies. Curr Opin Neurol. 2017;30:513-520. https://doi.org/10.1097/WCO.0000000000000475 PMid:28682959
- Gwathmey KG. Sensory neuronopathies. Muscle Nerve. 2016;53:8-19. https://doi.org/10.1002/mus.24943 PMid:26467754
- Graus
F, Ari-o H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin
and non-Hodgkin lymphomas. Blood. 2014;123:3230-3238. https://doi.org/10.1182/blood-2014-03-537506 PMid:24705493 PMCid:PMC4046430
- Briani
C, Vitaliani R, Grisold W, Honnorat J, Graus F, Antoine JC, Bertolini
G, Giometto B; PNS Euronetwork. Spectrum of paraneoplastic disease
associated with lymphoma. Neurology. 2011;76:705-710. https://doi.org/10.1212/WNL.0b013e31820d62eb PMid:21339498
- Smith
AG, Singleton JR. The diagnostic yield of a standardized approach to
idiopathic sensory-predominant neuropathy. Arch Intern Med.
2004;164:1021-1025. https://doi.org/10.1001/archinte.164.9.1021 PMid:15136313
- Zis
P, Sarrigiannis PG, Rao DG, Hewamadduma C, Hadjivassiliou M. Chronic
idiopathic axonal polyneuropathy: a systematic review. J Neurol.
2016;263:1903-1910. https://doi.org/10.1007/s00415-016-8082-7 PMid:26961897
- Sachchithanantham
S, Roussel M, Palladini G, Klersy C, Mahmood S, Venner CP, Gibbs S,
Gillmore J, Lachmann H, Hawkins PN, Jaccard A, Merlini G, Wechalekar
AD. European Collaborative Study Defining Clinical Profile Outcomes and
Novel Prognostic Criteria in Monoclonal Immunoglobulin M-Related Light
Chain Amyloidosis. J Clin Oncol. 2016;34:2037-2045. https://doi.org/10.1200/JCO.2015.63.3123 PMid:27114592
- Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007;36:411-423. https://doi.org/10.1002/mus.20821 PMid:17554795
- Lachmann
HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB,
Hawkins PN. Misdiagnosis of hereditary amyloidosis as AL (primary)
amyloidosis. N Engl J Med. 2002;346:1786-1791. https://doi.org/10.1056/NEJMoa013354 PMid:12050338
- Luigetti
M, Conte A, Del Grande A, Bisogni G, Madia F, Lo Monaco M, Laurenti L,
Obici L, Merlini G, Sabatelli M. TTR-related amyloid neuropathy:
clinical, electrophysiological and pathological findings in 15
unrelated patients. Neurol Sci. 2013;34:1057-1063. https://doi.org/10.1007/s10072-012-1105-y PMid:22592564
- Briani
C, Cavallaro T, Ferrari S, Taioli F, Calamelli S, Verga L, Adami F,
Fabrizi GM. Sporadic transthyretin amyloidosis with a novel TTR gene
mutation misdiagnosed as primary amyloidosis. J Neurol.
2012;259:2226-2228. https://doi.org/10.1007/s00415-012-6529-z PMid:22580845
- Coelho
T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V,
Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH, Trigo P,
Kelly JW, Labaudinière R, Chan J, Packman J, Wilson A, Grogan DR.
Tafamidis for transthyretin familial amyloid polyneuropathy: a
randomized, controlled trial. Neurology. 2012;79:785-792. https://doi.org/10.1212/WNL.0b013e3182661eb1 PMid:22843282 PMCid:PMC4098875
- Adams
D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV,
Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S,
Planté-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH
3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD,
Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur
SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB. Patisiran,
an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J
Med. 2018;379:11-21. https://doi.org/10.1056/NEJMoa1716153 PMid:29972753
- Benson
MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK,
Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M,
Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner
SB, Conceição I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD,
Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW,
Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T. Inotersen
Treatment for Patients with Hereditary Transthyretin Amyloidosis. N
Engl J Med. 2018;379:22-31. https://doi.org/10.1056/NEJMoa1716793 PMid:29972757
[TOP]