Maria Luisa Calabrò1* and Ronit Sarid2.
1 Immunology and Molecular Oncology, Veneto Institute of Oncology IOV - IRCCS, Padua, Italy
2
The Mina and Everard Goodman Faculty of Life Sciences & Advanced
Materials and Nanotechnology Institute, Bar-Ilan University, Ramat-Gan,
Israel.
Correspondence to: Dr. Maria Luisa Calabrò, Veneto Institute of
Oncology IOV – IRCCS, Immunology and Molecular Oncology, via
Gattamelata 64, 35128 Padua, Italy. Tel: +39-049-8215883. E-mail:
luisella.calabro@iov.veneto.it,
lcalabro@unipd.it
Published: November 1, 2018
Received: August 6, 2018
Accepted: October 2, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018061 DOI
10.4084/MJHID.2018.061
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
spectrum of lymphoproliferative disorders linked to human herpesvirus 8
(HHV-8) infection has constantly been increasing since the discovery of
its first etiologic association with primary effusion lymphoma (PEL).
PEL is a rapidly progressing non-Hodgkin’s B-cell lymphoma that
develops in body cavities in an effusional form. With the increase in
the overall survival of PEL patients, as well as the introduction of
HHV-8 surveillance in immunocompromised patients, the extracavitary,
solid counterpart of PEL was later identified. Moreover, virtually all
plasmablastic variants of multicentric Castleman’s disease (MCD)
developing in HIV-1-infected individuals harbor HHV-8, providing a
strong etiologic link between MCD and this oncogenic herpesvirus. Two
other pathologic conditions develop in HIV-1-infected persons
concomitantly with MCD: MCD with plasmablastic clusters and
HHV-8-positive diffuse large B-cell lymphoma not otherwise specified
(HHV-8+ DLBCL NOS), the first likely representing an intermediate stage
preceding the full neoplastic form. MCD in leukemic phase has also been
described, albeit much less commonly. The germinotropic
lymphoproliferative disorder (GLPD) may resemble extracavitary PEL, but
develops in immune competent HHV8-infected individuals, and, unlike the
other disorders, it responds well to conventional therapies. Almost all
HHV-8-mediated lymphoproliferative disorders are the result of an
interaction between HHV-8 infection and a dysregulated immunological
system, leading to the formation of inflammatory niches in which B
cells, at different developmental stages, are infected, proliferate and
may eventually shift from a polyclonal state to a monoclonal/neoplastic
disorder. Herein, we describe the association between HHV-8 and
lymphoproliferative disorders and highlight the predominant distinctive
features of each disease.
|
Introduction
Human
herpesvirus 8 (HHV-8) was first evidenced in 1993 in a Kaposi’s sarcoma
(KS) tissue sample by using representational difference analysis (RDA),
which enables unbiased detection of foreign DNA sequences in tumor
tissue as compared with its matched normal control.[1,2]
Four RDA fragments were detected, of which two were verified by
Southern blot analysis as unique to the diseased tissue. The amino acid
coding sequences of the newly identified KS-associated DNA shared a
high degree of homology with two known herpesviruses, Epstein-Barr
virus (EBV) and Herpesvirus Saimiri (HVS). Accordingly, the newly
identified human herpesvirus was termed “Kaposi’s sarcoma-associated
herpesvirus” (KSHV), and, later, HHV-8. KS-associated sequences were
subsequently detected in all epidemiological variants of KS, including
HIV-associated or epidemic, Mediterranean or classic, African or
endemic, and iatrogenic or post-transplant KS.[3-5] In
subsequent studies, HHV-8 sequences were detected in a lymphoma sample
from an AIDS patient which was initially included as a negative
control.[6] Then, a screening of 193 AIDS lymphoma
samples identified eight as positive for HHV-8 DNA; all were classified
as body cavity-based lymphoma (BCBL). This lymphoma, later designated
primary effusion lymphoma (PEL), represents an extremely rare
liquid-phase plasmablastic lymphoma which mostly appears in
HIV-1-infected patients. Its main characteristic is the large serous
cavities which constitute its site of primary development.[7]
Subsequently, the plasmablastic variant of multicentric Castleman’s
disease (MCD), which often presents with hyperplastic lymph nodes, was
linked to this herpesvirus,[8] in addition to two
proliferative disorders developing in patients with MCD: MCD with
plasmablastic aggregates and HHV-8-positive diffuse large B-cell
lymphoma not otherwise specified (HHV-8+ DLBCL NOS). An additional
condition that was later associated with HHV-8 infection and that
shares several characteristics with MCD is the KSHV-associated
inflammatory cytokine syndrome (KICS).[9] Like PEL and
MCD, this HHV-8-induced inflammatory condition develops at higher
frequencies in HIV-1-infected individuals. Conversely, the
germinotropic lymphoproliferative disorder (GLPD), which was described
later, develops mainly in HHV-8-infected immunocompetent hosts.[10]
HHV-8
has been classified by the International Agency for Research on Cancer
(IARC) as class I human carcinogen. The epidemiology, biology and
molecular characteristics of HHV-8 infection, as well as the potential
role of HHV-8 proteins and miRNAs in the pathogenesis of HHV-8, have
been widely reviewed.[11-14] Based on the Society of Hematopathology (SH)/European Association for Hematopathology 2015 Workshop Report,[15]
we describe in the present review the association between HHV-8 and
lymphoproliferative disorders and highlight the predominant distinctive
features of each disease, while indicating exceptions to the general
descriptions. Table 1 summarizes the pathological lymphoid conditions linked to HHV-8 infection and key features of these disorders.
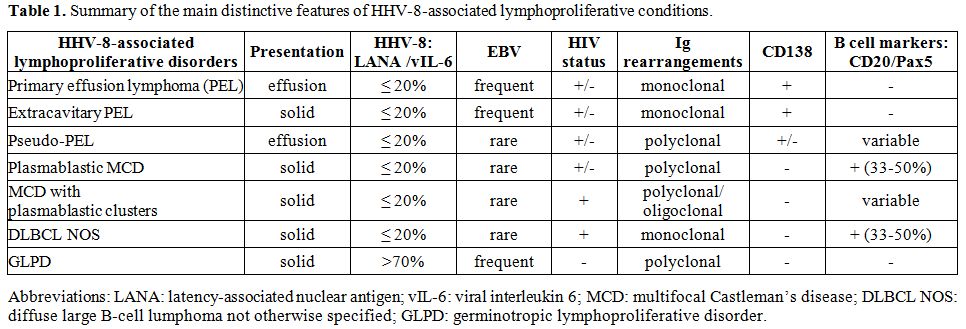 |
Table 1. Summary of the main distinctive features of HHV-8-associated lymphoproliferative conditions. |
Primary Effusion Lymphoma
Clinical and biological features.
PEL is a highly aggressive and rare non-Hodgkin’s B-cell lymphoma (NHL)
presenting as a liquid-phase lymphoma in body cavities, in particular,
the peritoneal, pericardial and pleural cavities.[7,16]
Symptoms are the result of the accumulation of neoplastic effusion and
patients with pleural or pericardial disease may present with dyspnea,
while those with peritoneal involvement with ascites. PEL may develop
concomitantly in more than one body cavity, and its prognosis is partly
linked to the number of involved intracavitary sites.[17]
Diagnostic criteria include: i) the presence of an effusional lymphoma,
with ii) monoclonal rearrangements of the immunoglobulin (Ig) variable
genes, and iii) detection of HHV-8 in the lymphomatous cells. Cytologic
preparations of the effusion show large neoplastic cells with round to
irregular nuclei and prominent nucleoli. The cells vary in appearance
from immunoblastic to plasmablastic and anaplastic, and numerous
mitotic figures are evident. Most PEL cells are latently infected with
HHV-8 and thus express only a small subset of viral genes, whereas a
small fraction of cells expresses viral lytic genes. Rarely, these
cells may harbor monoclonal rearrangements of T-cell receptor genes,
although in vivo and in vitro
studies have shown that, among the hematopoietic components, only B
lymphocytes and mononuclear cells can be infected with HHV-8.
PEL
cells have a peculiar immunophenotype as the lymphomatous cells do not
express classic B-cell (such as CD19, CD20 and PAX5) or T-cell (such as
CD3) lineage markers. They frequently express both activation (such as
CD38) and post-germinal center (GC) markers, such as MUM1/IRF4, B
lymphocyte-induced maturation protein 1 (Blimp-1) and the
characteristic adhesion molecule, Syndecan-1 or CD138.[18,19]
MUM1/IRF4 is a myeloma-associated transcriptionally active oncogene,
which is involved in the regulation of myc expression and B-cell
maturation and was found to be expressed in a high proportion of mature
lymphoproliferative disorders including B- and T-cell malignancies.[20,21]
Blimp-1 is a crucial transcriptional regulator, which is involved in
the terminal differentiation of B cells into plasma cells.
Interestingly, intracavitary targeting of Blimp-1 exerted a significant
anti-neoplastic effect in a preclinical SCID/PEL model, suggesting that
Blimp-1 represents a potential therapeutic target for PEL.[22]
Syndecan-1 is a cell-surface heparin-sulfate proteoglycan, generally
expressed on the basolateral surface of epithelial cells, and its
expression is correlated with cell differentiation and prognosis in
many types of tumors.[23] In the hematopoietic
compartment, this surface antigen is expressed at high density in
normal and transformed lymphocytes at the late stages of B-cell
differentiation.[24] The transcriptional profile of
PEL cells shows a pattern of gene expression intermediate between that
of a plasma cell and that of a diffuse large B-cell lymphoma.[25] Therefore, PEL cells seem to represent terminally differentiated, post-GC transformed B cells.
The
secretory profile of PEL cells includes high levels of viral and
cellular interleukin (IL) 6, IL-10 and vascular endothelial growth
factor (VEGF). Cellular and viral IL-6 (hIL-6 and vIL-6) promote B cell
growth and angiogenesis. hIL-6 was shown to be important for in vivo PEL cell proliferation.[26] IL-10 is one of the most important autocrine growth factors for PEL cells and is released by PEL cell lines at high levels in vitro, and throughout tumor progression in PEL murine models.[26-28]
The effect of VEGF, initially named vascular permeability factor, in
PEL pathogenesis was found to be mainly associated with the enhancement
of vascular permeability, thus contributing to the liquid growth of the
effusion rather than to neo-angiogenesis.[29]
Epidemiological subtypes.
Like KS, different epidemiological subtypes of PEL have been described.
The predominant variant is the one that develops in HIV-1-infected
individuals, in particular, advanced AIDS patients. In this population,
PEL represents about 4% of all HIV-associated NHLs whereas it accounts
for 0.3% of aggressive lymphomas developing in HIV-uninfected subjects.[30,31]
HIV-associated PEL develops more frequently in young male patients, and
has a very aggressive clinical course, with a median survival time of
2-6 months from diagnosis in the pre-antiretroviral therapy (ART)/early
combined ART (cART) era.[16,31,32]
Continuous cART therapy, along with high-dose chemotherapy regimens,
was found to ameliorate clinical aggressiveness by inducing, in certain
patients, a prolonged disease remission.[33,34] Of
note, PELs that are HIV-associated are frequently co-infected with EBV.
The “Mediterranean” or classic variant of PEL develops in HIV-negative
elderly patients, mostly in persons of Mediterranean basin descent.
This variant has an indolent clinical course and a more favorable
prognosis.[35-37] A post-transplantation PEL form has been described in renal, liver and cardiac transplant recipients.[38-40]
In these patients, PEL presents a variable clinical course, and it can
rapidly progress; removal of immunosuppressive therapy is often
associated with substantial clinical response. PEL can also develop in
HIV-negative subjects who are affected by Hepatitis C virus/Hepatitis B
virus-associated or cryptogenic liver cirrhosis.[41,42]
The African/endemic form of PEL remains to be identified, although its
existence is highly plausible. The lack of diagnosis or misdiagnosis of
African PEL patients could be primarily explained by difficulties in
performing appropriate viral, histopathological and instrumental
analyses in this population. Moreover, these patients are frequently
affected by several comorbidities, further complicating the diagnostic
procedure.
Pathogenic mechanisms.
The initial step that promotes PEL onset is common to all disorders
that are linked to HHV-8 infection, i.e., immune activation leading to
increased inflammatory cytokine secretion (reviewed in Ensoli et al.).[43]
This condition favors the lytic program of HHV-8 infection and
increases the pool of infected cells in the systemic compartment, which
in turn amplifies the inflammatory profile. Of note, effusions composed
of polyclonal atypical HHV-8-infected B cells that are surrounded by an
inflammatory microenvironment have been documented in body cavities of
HIV-infected subjects who are affected by other HHV-8-associated
disorders. Given the lack of tumor monoclonality and the discrepancy
between viral and cellular clonality in a number of effusion samples, a
possible transition phase towards HHV-8-positive PEL has been
suggested.[44,45]
Inflammatory cytokines are
also thought to be responsible for the activation of mesothelial cells,
representing specialized epithelioid cells lining the body cavities.[46]
In response to an injury or to altered intracavitary homeostasis,
mesothelial cells may themselves release inflammatory mediators which
recruit leukocytes from the systemic compartment through a chemotactic
gradient and by the exposure of adhesion molecules and integrins on the
mesothelial cell surface. Accordingly, mesothelial cells amplify and
extend the pattern of cytokines through the secretion of growth factors
and chemokines, thereby recruiting potentially infected B cells to
intracavitary sites. Like the activated foci of the endothelial cells
in KS pathogenesis,[47] this cytokine-rich
microenvironment may favor homing, and possibly, the proliferation of
PEL precursors in the intracavitary compartment, eventually leading to
their oncogenic conversion.
Nevertheless, mesothelial cells also
function as guardians of the intracavitary homeostasis and the first
line of defense against infections,[48] and may thus
assist in the resolution of the inflammatory wave. A recent study
hypothesized that PEL cells might derive from mesothelial cells lining
body cavities, through a mesothelial-lymphoid transition, a biological
process likely responsible for the genesis of resident B1 lymphocytes.[49]
This hypothesis is in line with the plasticity of mesothelial cells.
However, it does not explain the immunophenotypic and genetic features
of PEL cells in vivo.
In
the absence of therapy, PEL is rapidly progressive and consistently
lethal. The rapid progression of this lymphoma is linked to its
peculiar site of development. Indeed, in vitro
co-culture studies showed that mesothelial cells could modify the
turnover of PEL cells by increasing their proliferation and their
resistance to apoptotic stimuli, thus generating an environment
favorable to PEL progression.[50] On the other hand,
PEL cells, through the release of Transforming Growth Factor
(TGF)-beta, induce type 2 epithelial-mesenchymal transition (EMT) in
primary human mesothelial cells.[50,51] This can be
observed in co-culture systems as well as in a preclinical animal model
of SCID/PEL mimicking the aggressive nature of PEL in humans.[28]
Primary human mesothelial cells have a polygonal shape and form a
regular monolayer in culture. Their co-culture with PEL cells, or
treatment with TGF-beta, induces morphological changes within a few
days which lead to a spindle-like shape, with a myofibroblastic
morphology, and formation of small foci with accumulation of elongated
mesothelial cells (Figure 1).
These morphological changes are accompanied by transcriptional
re-programming characterized by up-regulation of specific transcription
factors (Snail, Slug, Zeb1 and Sip1) that lead to downregulation of
E-cadherin and other proteins mediating cell-to-cell contacts, in
particular, adherens and tight junctions. Some structural proteins are
also substituted by other cytoskeletal components that facilitate cell
motility, such as α-SMA. This conversion into a myofibroblast-like
phenotype can also be visualized during ascites development in mice
intraperitoneally injected with PEL-derived cell lines. Indeed, the
occurrence of EMT in vivo was
demonstrated through the thickening of the mesothelium lining the
peritoneal cavity during ascites progression in a PEL preclinical model
(Figure 2, panels A and B).[50] Serosal thickening was accompanied by loss of cytokeratin staining, both of which are typical signs of fibrosis.[52] Interestingly, this phenomenon was also documented in PEL patients,[53] indicating that fibrosis occurs during PEL progression. Type 2 EMT was shown to increase the survival of PEL cells,[50]
suggesting that this phenomenon in the intracavitary microenvironment
may be responsible for the aggressive nature of PEL. Moreover,
discontinuity in the mesothelial layer might also contribute to the
occurrence of extracavitary PEL.
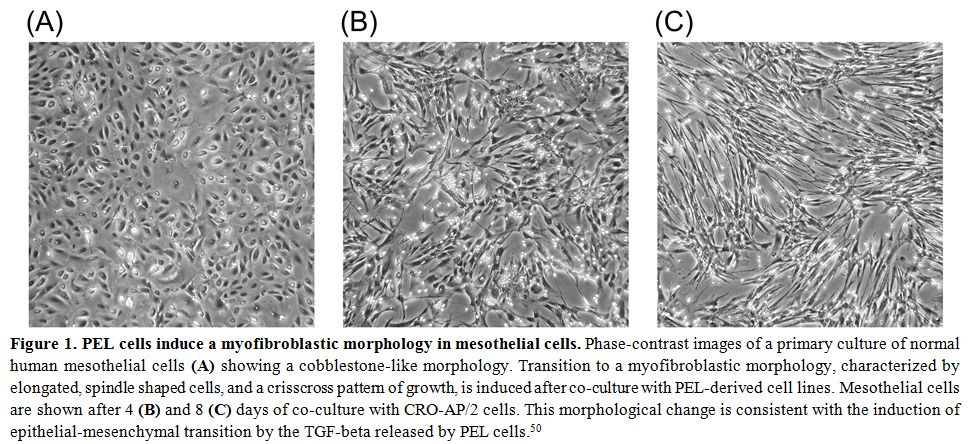 |
Figure 1. PEL cells induce a myofibroblastic morphology in mesothelial cells.
Phase-contrast images of a primary culture of normal human mesothelial
cells (A) showing a cobblestone-like morphology. Transition to a
myofibroblastic morphology, characterized by elongated, spindle shaped
cells, and a crisscross pattern of growth, is induced after co-culture
with PEL-derived cell lines. Mesothelial cells are shown after 4 (B)
and 8 (C) days of co-culture with CRO-AP/2 cells. This morphological
change is consistent with the induction of epithelial-mesenchymal
transition by the TGF-beta released by PEL cells.[50] |
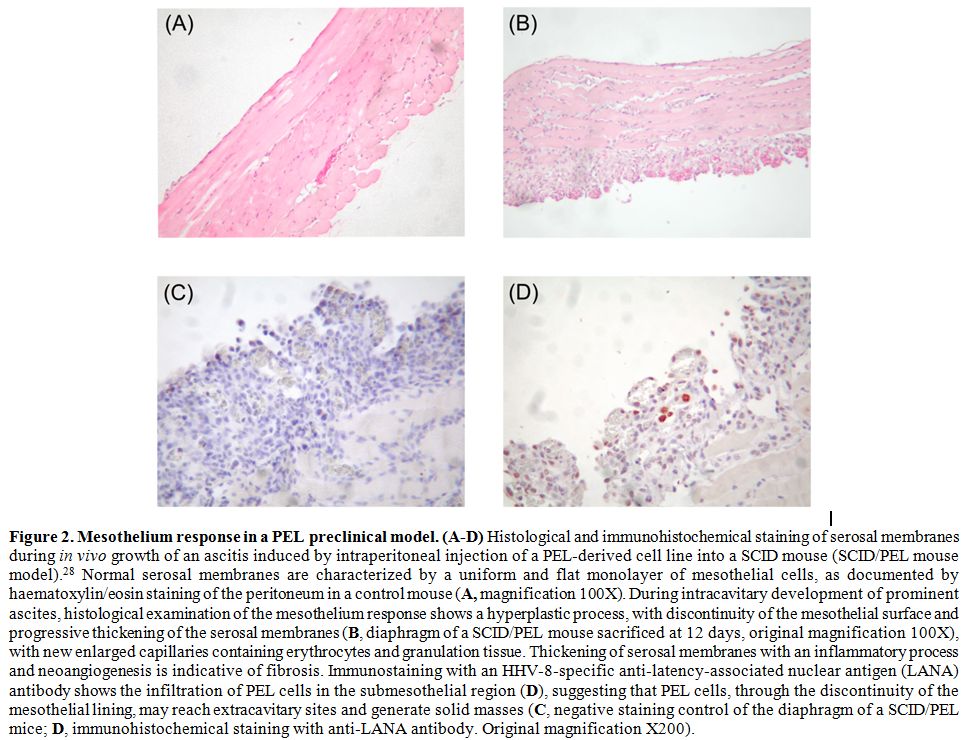 |
Figure 2. Mesothelium
response in a PEL preclinical model. (A-D) Histological and
immunohistochemical staining of serosal membranes during in vivo growth
of an ascitis induced by intraperitoneal injection of a PEL-derived
cell line into a SCID mouse (SCID/PEL mouse model).28 Normal serosal
membranes are characterized by a uniform and flat monolayer of
mesothelial cells, as documented by haematoxylin/eosin staining of the
peritoneum in a control mouse (A,
magnification 100X). During intracavitary development of prominent
ascites, histological examination of the mesothelium response shows a
hyperplastic process, with discontinuity of the mesothelial surface and
progressive thickening of the serosal membranes (B,
diaphragm of a SCID/PEL mouse sacrificed at 12 days, original
magnification 100X), with new enlarged capillaries containing
erythrocytes and granulation tissue. Thickening of serosal membranes
with an inflammatory process and neoangiogenesis is indicative of
fibrosis. Immunostaining with an HHV-8-specific anti-latency-associated
nuclear antigen (LANA) antibody shows the infiltration of PEL cells in
the submesothelial region (D), suggesting that PEL cells, through the
discontinuity of the mesothelial lining, may reach extracavitary sites
and generate solid masses (C,
negative staining control of the diaphragm of a SCID/PEL mice; D,
immunohistochemical staining with anti-LANA antibody. Original
magnification X200). |
Extracavitary/solid PEL
Clinical and biological features. The extracavitary variant of PEL was reported after the description of the classic effusion form of PEL.[54,55] Extracavitary PEL more frequently develops after or concomitantly with the emergence of the effusion form;[56-58] however, it may, albeit more rarely, precede this form.[54,59,60] Cases of solid PEL that are not accompanied by the effusional form have also been described.[54]
Lymphomatous
cells of solid PEL frequently share similar morphological,
immunophenotypic, genotypic and virologic characteristics with those
described for the effusional form. However, this lymphoma consists of a
solid mass that may be present at different organs. This variant of PEL
may exclusively involve the lymph nodes, or affect other organs,
including the gastrointestinal tract, lungs, skin, and the central
nervous system. Extracavitary PEL develops more frequently in
HIV-infected subjects and, recently, two cases of African extracavitary
PEL have been described.[61] However, cases in HIV-negative individuals have also been reported.[58]
Of
note, HIV/HHV-8-infected subjects may also concomitantly develop other
HHV-8-positive DLBCL, as described below, characterized by
immunoblastic or immunoblastic/plasmacytoid cells, which have different
characteristics but share HHV-8 infection. Therefore, solid PEL is much
more challenging to diagnose as compared to its effusion counterpart.
Detection of HHV-8 in the lymphomatous cells can solve some ambiguous
cases, although it might not be the primary diagnostic approach in
cases of solid lymphomas. Lymphomatous cells have a variable
morphology, ranging from plasmablastic to immunoblastic and anaplastic,
but they generally lack B-cell markers, as well as surface and
cytoplasmic immunoglobulins. Diagnostic criteria include detection of
HHV-8 in tumor cells, frequent detection of CD138 and CD38, infrequent
staining for B-cell markers, and presence of monoclonal IgH gene
rearrangements (Table 1). Concomitant intracavitary effusion might also support the diagnosis.
Chadburn et al.[54]
demonstrated a longer survival of extracavitary PEL patients compared
to classic PEL patients, with a median survival time of 11 versus 3
months, respectively. Another study compared the characteristics and
the outcome of classic PEL with those of solid PEL in HIV-1-infected
patients receiving combined antiretroviral therapy.[34]
Similar clinical, morphological and immunophenotypic characteristics
were reported in the two groups, with a similar high frequency
(>59%) of EBV detection. The majority of patients received standard
chemotherapy with cyclophosphamide, doxorubicin, vincristine and
prednisone (CHOP-based) combined with methotrexate. Complete remission
was achieved in 62% of the patients with the classic variant, and in
41% of the patients affected by the extracavitary form. The median
overall survival was similar in the two groups (10.2 months),
regardless of the treatment regimen; yet, it is interesting to note
that, in accordance with other studies,[54] patients
with the extracavitary localization, who achieved initial remission,
remained disease-free during a 10-year follow-up, whereas more than
half of the surviving patients with the effusional PEL relapsed.
Pathogenetic mechanisms.
Most PEL patients either present with coexisting solid and effusional
forms of PEL or develop a solid mass following the diagnosis of a
liquid-phase PEL.[54,58,62]
Concerning the possible pathogenic mechanisms involved in the
development of this PEL variant, it was proposed that type 2 EMT
involving the mesothelium lining the body cavities during effusional
PEL progression might favor the subsequent or concomitant formation of
an extracavitary mass.[50] This mechanism is supported by the frequent finding of lymphocyte infiltration of the serosal membranes in PEL patients.[56]
Extracavitary PEL is more frequent during the course of intracavitary
effusions and likely reflects the occurrence of serosal discontinuity
and consequent leakage of lymphomatous cells. Due to the rapid
progression of PEL, it is conceivable that the occurrence of the
extracavitary migration of lymphomatous PEL cells would be mainly
observed in longer-surviving patients. Indeed, extracavitary secondary
masses were also described in PEL patients after treatment.[32,55]
Actually, these patients experienced PEL remission and relatively
longer survival, with a disease-free interval ranging from 5 to 97
months. In one case, the extracavitary mass, which developed following
a PEL-free interval, was morphologically and immunophenotypically
different from the initial PEL tumor and was compatible with the
occurrence of a distinct HHV-8-associated MCD-associated oligoclonal
“microlymphoma”[55] (see below). In other cases,
however, the extracavitary mass and the primary PEL had similar
morphological characteristics, immunophenotypic profile and clonal
identity, thus providing evidence that the extracavitary solid mass
represents the relapse of the original effusional lymphoma at a
different site, as a solid tumor.[55,58] Interestingly, this finding was also reproduced in a preclinical SCID/PEL model after treatment [(Figure 2)[28]
and unpublished data]. The seeding of tumor cells through the
submesothelial layer to form a solid mass is also supported by studies
in mice treated with interferon (IFN)-alpha. Mice intraperitoneally
injected with PEL cells and transduced with a lentiviral vector
expressing IFN-alpha showed significantly increased survival compared
to control mice, along with a significant reduction in ascites
development. These mice subsequently developed extracavitary masses,[28]
suggesting that PEL cells could leave the intracavitary site during
initial ascites formation, and thus escape the intracavitary treatment
to generate extra-peritoneal solid masses later.
However, PEL
patients presenting exclusively with a solid PEL, or having a solid PEL
followed by a liquid-phase PEL have also been described,[54,60,63,64]
thereby suggesting a different pathogenic mechanism. As these solid
PELs occurred mainly in the gastrointestinal tract and in the skin, it
has been hypothesized that solid PEL occurs at the same sites of KS
development. This hypothesis should imply that the same pathogenic
mechanisms leading to the transformation of lymphatic endothelial cells
into spindle cells following HHV-8 infection may also apply, though
rarely, to B cells recruited to these anatomic sites.
Pseudo-PEL
Patients affected by MCD and/or KS may develop recurrent non-lymphomatous effusions in body cavities.[44,45,65]
These effusions show certain features of PEL, including HHV-8
infection, generally with high cell-associated viral loads, the
involvement of one or more body cavities, and aggressive clinical
course. Moreover, they may arise in both HIV-negative and HIV-positive
patients.
However, unlike PEL, in these lymphoproliferative
disorders, the fraction of HHV-8-infected cells is small, and the
infected cells are atypical lympho-mononuclear cells which commonly
express the latency-associated nuclear antigen (LANA) and vIL-6. No
evidence for T-cell/B-cell clonality is generally found. Certain cases
may show monotypic (IgM/ λ)
LANA-positive lymphoid cells resembling the plasmablasts of MCD; more
rarely, cases of LANA+/CD68R+ mononuclear cells on an inflammatory
background may be reminiscent of an effusive form of an early
polyclonal KS lesion. These polyclonal HHV-8-positive effusions may,
therefore, represent an early phase of PEL or a liquid form of MCD or
KS. Conversely, they could represent a distinct HHV-8-associated
inflammatory, infectious process arising in body cavities.
Multicentric Castleman’s Disease (MCD)
Clinical and biological features.
MCD is a rare polyclonal B-cell lymphoproliferative disorder,
presenting as generalized lymphadenopathy with angiofollicular
hyperplasia and affecting both HIV-positive and negative subjects.
Castleman’s disease, first described in 1956,[66] is
characterized by a benign mass of lymphoid tissue that may involve a
single lymph node (“unicentric”) with an estimated incidence of 16 per
million person years, or several lymph nodes (“multicentric”), with a
lower incidence of 5 per million person years.[67-69]
Three variants have been described based on their histopathological
features. These include: i) the hyaline vascular form, which is
characterized by a vascular lymphoid tissue containing lymphocytes
forming ordered, concentric “onion skin” layers around the large
follicles, with frequent perivascular hyalinization; ii) the
plasmablastic variant, which shows large sheets of plasma cells
expanding the mantle zone and generally preserved architecture of lymph
nodes, and iii) the mixed variant, which shows both histological
patterns. The
observation that more than 50% of AIDS-KS patients are also affected by
MCD prompted the search for HHV-8 sequences in MCD in both HIV-infected
and -uninfected subjects. After that, HHV-8 was detected in
plasmablastic and mixed variants.[8] Within affected
lymph nodes, HHV-8 is detected in IgM-positive plasmablasts. The
immunophenotype of HHV-8-infected cells is not strictly plasmablastic,
as these cells may be, more rarely, positive for CD20, and frequently
negative for CD138 (Table 1).[70,71]
All plasmablasts in lymph nodes and spleen express LANA, along with
MUM1/IRF4 and PAX5, and are Bcl-6-negative. PAX5 is a transcription
factor which participates in the maintenance of the B-cell lineage;[72]
its protein expression level, as detected by IHC, is used to assess the
B-cell lineage. HHV-8-infected plasmablasts are polyclonal but have a
restricted monotypic (IgM-lambda) phenotype.[73] They
do not harbor somatic mutations in the rearranged Ig genes, indicating
their origin from naïve pre-GC B cells. Concomitant EBV infection is
found very rarely. HHV-8 is involved in 60-100% of HIV-associated MCD
cases, and in 20-40% of HIV-negative patients.[74-77] Formal criteria have been established for the diagnosis of HHV-8-unrelated/idiopathic MCD.[78] HHV-8
infected B plasmablasts in lymph nodes produce high levels of viral and
cellular IL-6, which are thought to be responsible for the systemic
illness, and what are known as “B” symptoms, including severe fatigue,
fever, edema, weight loss, cell-free pleural effusion, anemia,
thrombocytopenia, lymphadenopathy and splenomegaly.[79,80]
Lymph node resection in MCD patients was found to reduce both symptoms
and hIL-6 serum levels, indicating that hIL-6 is mainly produced within
the germinal center of the involved lymph nodes and has a major role in
the pathogenesis of the disease.[81] Moreover,
transgenic mice that constitutively express murine IL-6 or vIL-6 were
shown to develop an MCD-like syndrome, further supporting the
influential role played by this cytokine.[82,83] The
symptomatic phase is very frequently accompanied by elevated levels of
cell-free HHV-8 viremia, generally higher than those reached in
patients affected by other HHV-8-linked pathologies. Rare
cases of MCD with the presence of plasmablasts in the systemic
compartment have been reported. These cases of MCD in leukemic phase
were described in HIV-infected subjects with a history of KS in the
presence[84] or absence[85] of a diffused lymphoadenopathy. The
criteria to distinguish HHV-8-associated MCD from lymphoid hyperplasia
may principally rely on the constant presence of HHV-8 in the
plasmablasts. Apart from being PCR-positive for different genomic
regions of the virus, plasmablasts generally express LANA and also
express vIL-6 in a small fraction (generally < 20%) of the cells (Table 1).
A similar viral expression pattern has also been observed in PEL and in
pseudo-PEL cells. Lymphocyte proliferation is generally polyclonal.8
However, MCD can progress to HHV-8+ DLBCL NOS or to a form presenting
aggregates of plasmablastic cells, which likely represent an
intermediate stage preceding the frank monoclonal disease.[8,71] Pathogenic mechanisms.
Increased loads of HHV-8, together with increased IL-6 and IL-10
levels, exacerbate disease in patients with MCD. Therefore, a model
that considers these factors in the pathogenesis of the disease has
been proposed,[86] that may explain the pathogenic role of HHV-8 in the
three variants of MCD. This model relies on similarities between KS and
MCD in multiple characteristics including HHV-8 infection and frequent
association with HIV dysregulated production of human and viral IL-6,
the inflammatory profile, histopathological characteristics and initial
polyclonal proliferation that can progress to a monoclonal neoplastic
process. The scenario is the lymph node, in which inflammatory
cytokines boost the lytic cycle of HHV-8 and the subsequent viral
dissemination to B lymphocytes and lymphovascular endothelial cells.
This process is amplified in the presence of HIV infection, which
increases the level of inflammation and cooperates directly and
indirectly to increase HHV-8 loads.Moreover,
prominent lytic viral replication leads to hyalinization, which also
characterizes KS, and results in the destruction of lymphovascular
endothelial cells. This “two-compartment” propagation might ultimately
explain the simultaneous presence of KS and MCD in HIV/HHV-8-coinfected
individuals.[86] However, this complex interplay
might also give rise to heterogeneous clinical and histopathological
outcomes controlled by the ratios between cellular and viral cytokines,
and between the lytic and latent programs. Indeed, this model could
explain not only the two pathologies associated with MCD (plasmablastic
aggregates and HHV-8+ DLBCL NOS) but also the extracavitary PEL,
characterized by viral infection and monoclonal expansion of a
different subset of post-GC B cells that is promoted by the
inflammatory microenvironment.
MCD with Plasmablastic Aggregates
MCD
can progress towards a transitional polyclonal or, more rarely,
oligoclonal entity of microscopic dimensions. This entity,
characterized by HHV-8 infection and presence of clusters of
LANA-expressing blasts colonizing or substituting the germinal centers
of lymph nodes, was initially designated foci of “microlymphoma”.[71,73]
Due to the absence of monoclonality and because a full-blown lymphoma
does not always develop, this term is no longer used. Nevertheless,
such aggregates may precede the subsequent development of a frank
monoclonal HHV-8-positive plasmablastic lymphoma, more recently
designated HHV-8+ DLBCL NOS.[15] Like
MCD, this disorder is more frequent in HIV-infected subjects, in
concordance with other HHV-8-associated disorders, and plasmablasts may
also express vIL-6, along with IRF4/MUM1.[15] EBV co-infection is rare but has been described.[87]Thus,
histopathological and molecular analyses of MCD lymph nodes show that
HHV-8-infected plasmablasts may form microscopic sheets or clusters of
large polyclonal cells with a restricted monotypic (IgM-λ) phenotype, without somatic mutations in the rearranged immunoglobulin gene, supporting their origin from naïve pre-GC B cells.
HHV-8+ Diffuse Large B-Cell Lymphoma Not Otherwise Specified (HHV-8+ DLBCL NOS)
DLBCL
represents one of the most frequent types of B-cell lymphomas and may
encompass different morphological subtypes, including the plasmablastic
lymphoma, originally described in the oral cavity of HIV-1-infected
individuals.[88] HHV-8+ DLBCL NOS usually develops in MCD patients in the context of HIV infection.[8,71,89]
Histopathological examination of lymph nodes and spleen reveals that
their architecture is severely damaged and replaced by large sheets of
malignant monoclonal cells. Extranodal sites may also be affected.
Lymphomatous cells have a plasmablastic or, less frequently, an
immunoblastic morphology. HHV-8+ DLBCL NOS has an immunophenotypic
pattern characterized by lack of B-cell markers, expression of λ and, to a lesser extent, κ light chains, and presence of plasma cell markers (CD38+, IRF4/MUM1+).[90]
EBV co-infection of plasmablasts is extremely rare. This lymphoma can
be predicted by the presence of polyclonal/oligoclonal aggregates of
plasmablastic cells, as described in the previous section.Lymphoma
with plasmablastic differentiation found in HIV-1-infected subjects
might have quite heterogeneous features and may be distinguished on the
basis of differential antigen expression and association with viruses.[89,91-93]
Differential diagnosis between HHV-8+ DLBCL NOS and extracavitary PEL
can be performed on the basis of CD138 and EBV, which are frequently
found in intracavitary and solid forms of PEL (Table 1).
PEL cells are post-GC B cells that have undergone an intense somatic
mutation process on the immunoglobulin gene hypervariable region.
Conversely, lymphomatous cells of HHV-8+ DLBCL NOS are naïve,
non-mutated, pre-GC B cells.
KSHV-Associated Inflammatory Cytokine Syndrome (KICS)
The KSHV-associated inflammatory cytokine syndrome is not a lymphoproliferative disorder per se
but has several clinical, radiologic and virologic similarities with
MCD. It was proposed as a unique clinical condition with a high
mortality rate associated with HHV-8 infection.[9,94]
The six initially described patients (five of them HIV-infected)
presented with a severe systemic inflammatory syndrome
indistinguishable from that found in the symptomatic phase of
HHV-8-associated plasmablastic MCD, characterized by high HHV-8 viremia
and elevated systemic levels of IL-6, vIL-6 and IL-10. Besides the
similar clinical presentation, histopathological studies could not
document nodal signs of MCD in any of the patients. Moreover KICS was
shown to occur concurrently with other HHV-8-associated disorders,
specifically KS or PEL.[9,94] It has
been hypothesized that this condition may precede the development of
HHV-8-associated disorders, although most KICS cases have been
described in patients presenting with other, already developed,
HHV-8-related lymphoproliferative diseases. KICS-like inflammatory manifestation with the HHV-8 association has also been described in transplant recipients.[95-97] Of note, a clinical case of kidney-liver post-transplant KICS was recently reported.[97]
This transplant patient presented with unexplained fever, markers of
severe systemic inflammation including increased IL-6, IL-10, IL-8, and
granulocyte-macrophage colony-stimulating factor (GM-CSF) plasma
levels, and elevated HHV-8 viremia. This patient, who was HIV-negative,
was found to have a donor-derived primary HHV-8 infection and was
successfully treated by a combination of antivirals, anti-CD20
monoclonal therapy, and modulation of the immunosuppressive regimen.[97]
Accordingly, it appears that KICS is an underestimated condition that
should be carefully monitored in the transplant setting.
Germinotropic Lymphoproliferative Disorder (GLPD)
This
lymphoproliferative disorder was first identified in 2002 in three
immunocompetent subjects presenting with a localized lymphoadenopathy
and responding satisfactorily to conventional therapy.[10]
Histopathological examination of the lymphoadenopathy detects
plasmablasts which preferentially invade the germinal centers of
follicles; the overall structure of the lymph node generally remains
undamaged. Plasmablasts are polyclonal or oligoclonal. Virological
studies showed that plasmablasts are very frequently coinfected with
both HHV-8 and EBV. In
contrast to PEL and MCD-associated plasmablasts, in which the
percentage of LANA-positive cells that also express vIL-6 is generally
lower than 20%, GLPD nodes contain a large fraction of
LANA/vIL-6-positive cells (Table 1).
These cells present latency I phenotype for EBV, being negative for
LMP1, EBNA2, and BZLF-1 expression. They may present in clusters as
those described in HIV-infected subjects affected by MCD, but these
plasmablasts are CD20-/CD27-/CD138-/CD10-/CD79a-/Bcl-6-/Bcl-2. Less
than ten canonical GLPD cases have been described to date, all in
immunocompetent subjects.[98]
Principal distinctive features for differential diagnosis
Table 1
summarizes the principal distinguishing features of HHV-8-associated
lymphoproliferative disorders, which may require differential
diagnosis. Diagnosis of classic PEL is mainly based on radiological
evidence of a malignant effusion involving the pleural, pericardial or
peritoneal cavities, with or without a tumor mass, predominantly
composed of monoclonal lymphomatous cells that are infected with HHV-8.
HHV-8 infection can be easily demonstrated by LANA staining by
immunohistochemistry or immunofluorescence of the neoplastic cells. Plasmablastic
or immunoblastic solid lymphoma with LANA-expressing cells, which could
also appear concomitantly with liquid-phase PEL, may represent
extracavitary localization of PEL but could also be an HHV-8+ DLBCL
NOS. Although both neoplasms may present a small fraction of
LANA/vIL-6-coexpressing lymphomatous cells, differential diagnosis may
be based on immunophenotype with the presence of CD138 expression and
absence of conventional B-cell markers in the extracavitary PEL, and
the opposite staining pattern in MCD-associated diffuse large B-cell
lymphoma NOS (Table 1). Another
important distinguishing feature is the frequent presence of EBV
co-infection in the extracavitary PEL, whereas this herpesvirus is very
rarely detected in HHV-8-induced MCD-associated lymphoproliferative
manifestations. In
HIV-negative subjects, a localized or diffuse lymphoadenopathy may
manifest the clinical presentation of extracavitary PEL, plasmablastic
MCD or GLPD. Extracavitary PEL and GLPD are frequently co-infected with
EBV, whereas this herpesvirus is very rarely detected in plasmablastic
MCD. The high percentage of cells co-expressing LANA and vIL-6
(>70%) should favor the diagnosis of GLPD rather than plasmablastic
MCD. Both conditions show a polyclonal pattern and lack of Syndecan-1
expression. These last two features should exclude the diagnosis of an
extracavitary PEL. It
must be noted that, while these features may help in differential
diagnosis, several exceptions to these general characteristics have
been reported in the literature. Actually, HHV-8-associated lymphomas
may be highly heterogeneous.[15] Indeed, cases of
liquid-phase or extracavitary PEL expressing T-cell markers (CD3, CD2,
CD5 or CD7) or B-cell markers (CD19, CD20, CD23, CD79a) have been
described.[16,34,99,100]
The rare cases of PEL expressing T-cell markers, such as CD3 and CD4,
presumably derive from the coexistence of B and T cell clones.
Moreover, cases of HHV-8-positive malignant effusions in body cavities
other than the three main sites have also been reported, such as those
arising in body cavities surrounding breast implants and those
involving the cerebrospinal fluid.[101,102]
Malignant cells resembling plasmablasts characterize HHV-8+ DLBCL NOS,
but they may also have immunoblastic morphology. They are monoclonal,
usually expressing the λ light chain, but cases expressing the κ light chain have been also reported.[89]Moreover,
positivity for B-cell markers, such as CD20, may vary from 30 to 50%,
and CD79a may be absent, as in PEL cells. GLPD is characterized by
large atypical cells, with plasmablastic, anaplastic or immunoblastic
features, usually negative for CD20 and CD138, but positive for
IRF4/MUM1 with monotypic κ or λ
light chain expression. However, cases not expressing monotypic light
chains or cases expressing CD38 or CD138 have also been reported.[103]
Moreover, a few cases with features transitioning between MCD with
plasmablastic aggregates and GLPD have been described, as were cases
developed in HIV-infected subjects.[93,103]
All together, these findings indicate that the spectrum of
HHV-8-associated lymphoproliferative disorders may be wider than that
described to date.
Conclusions
Although HHV-8 can infect several cell types in vitro, its tropism in vivo
appears to be restricted to two main cellular targets: the
lymphovascular endothelium and B lymphocytes. An inflammatory condition
is the primum movens of the
dynamic processes that lead to the neoplastic conversion of these two
cell types. The oncogenic process involves activation of the lytic
cycle of HHV-8, which in turn increases the amount of circulating
infected cells and promotes virus dissemination to other tissues. In
addition, cytokine-rich niches are responsible for the boost of the
polyclonal proliferation of B cells, in the case of lymphoproliferative
disorders, and/or of endothelial cells, in the case of KS, at different
body sites, including lymph nodes, dermis and body cavities. In this
scenario, the HHV-8-infected B cell and the inflammatory niches are key
drivers of the variegated histopathological and immunophenotypic
characteristics of HHV-8-associated lymphoproliferative disorders.
Moreover,
in HIV-infected individuals, these processes are highly amplified,
since HIV-infected cells augment the level of inflammatory cytokines,
and HIV viral products directly cooperate in the activation of the
lytic cycle of HHV-8. This is supported by the evidence that
HIV/HHV-8-coinfected individuals may experience two, or even more,
HHV-8-linked disorders, sequentially or concurrently. Other patients
with immunological dysfunctions may develop these disorders, and
transplant recipients should be carefully and frequently examined for
HHV-8-induced neoplastic and non-neoplastic conditions. Nevertheless,
HHV-8-infected individuals acquiring HIV-1 infection are those with the
highest risk for HHV-8-associated lymphoproliferative disorders. Search
for HHV-8 should be therefore routinely performed in all cases of
proliferative lymphoid disease arising in HIV-infected patients,
particularly in the absence of a robust immunovirological response to
cART.
Aknowledgements
This study was supported by 5X1000-IOV2011 (grant CUP n. J94G14000180001 to MLC).
References
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J,
Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences
in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865-9. https://doi.org/10.1126/science.7997879 PMid:7997879
- Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946-51. https://doi.org/10.1126/science.8438152 PMid:8438152
- Moore
PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi's
sarcoma in patients with and those without HIV infection. N Engl J Med.
1995;332:1181-5. https://doi.org/10.1056/NEJM199505043321801
- Huang
YQ, Li JJ, Kaplan MH, Poiesz B, Katabira E, Zhang WC, Feiner D,
Friedman-Kien AE. Human herpesvirus-like nucleic acid in various forms
of Kaposi's sarcoma. Lancet. 1995;345:759-61. https://doi.org/10.1016/S0140-6736(95)90641-X
- Dupin
N, Grandadam M, Calvez V, Gorin I, Aubin JT, Havard S, Lamy F,
Leibowitch M, Huraux JM, Escande JP, et al. Herpesvirus-like DNA
sequences in patients with Mediterranean Kaposi's sarcoma. Lancet.
1995;345:761-2. https://doi.org/10.1016/S0140-6736(95)90642-8
- Chang Y, Moore P. Twenty years of KSHV. Viruses. 2014;6:4258-64.10.3390/v6114258. https://doi.org/10.3390/v6114258
- Cesarman
E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated
herpesvirus-like DNA sequences in AIDS-related body-cavity-based
lymphomas. N Engl J Med. 1995;332:1186-91. https://doi.org/10.1056/NEJM199505043321802
- Soulier
J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P,
d'Agay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi's
sarcoma-associated herpesvirus-like DNA sequences in multicentric
Castleman's disease. Blood. 1995;86:1276-80. http://www.ncbi.nlm.nih.gov/pubmed/7632932 PMid:7632932
- Uldrick
TS, Wang V, O'Mahony D, Aleman K, Wyvill KM, Marshall V, Steinberg SM,
Pittaluga S, Maric I, Whitby D, Tosato G, Little RF, Yarchoan R. An
interleukin-6-related systemic inflammatory syndrome in patients
co-infected with Kaposi sarcoma-associated herpesvirus and HIV but
without Multicentric Castleman disease.Clin Infect Dis.
2010;51:350-8.10.1086/654798. https://doi.org/10.1086/654798
- Du
MQ, Diss TC, Liu H, Ye H, Hamoudi RA, Cabecadas J, Dong HY, Harris NL,
Chan JK, Rees JW, Dogan A, Isaacson PG. KSHV- and EBV-associated
germinotropic lymphoproliferative disorder. Blood. 2002;100:3415-8. https://doi.org/10.1182/blood-2002-02-0487
- Mesri EA, Cesarman E, Boshoff C. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10:707-19. https://doi.org/10.1038/nrc2888
- Purushothaman P, Uppal T, Verma SC. Molecular biology of KSHV lytic reactivation. Viruses. 2015;7:116-53. https://doi.org/10.3390/v7010116
- Schulz TF, Cesarman E. Kaposi Sarcoma-associated Herpesvirus: mechanisms of oncogenesis. Curr Opin Virol. 2015;14:116-28. https://doi.org/10.1016/j.coviro.2015.08.016
- Sarid
R, Calabrò ML. Kaposi's Sarcoma-Associated Herpesvirus: Epidemiology,
Biological Characteristics and Pathogenesis. In: Kaslow R, Stanberry
LR, Le Duc JW eds. Viral Infections of Humans: Epidemiology and
Control. 5th ed., New York: Springer. 2014:897-931.
- Chadburn
A, Said J, Gratzinger D, Chan JK, de Jong D, Jaffe ES, Natkunam Y,
Goodlad JR. HHV8 / KSHV-Positive Lymphoproliferative Disorders and the
Spectrum of Plasmablastic and Plasma Cell Neoplasms: 2015 SH/EAHP
Workshop Report-Part 3. Am J Clin Pathol. 2017;147:171-87. https://doi.org/10.1093/ajcp/aqw218
- Nador
RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM.
Primary effusion lymphoma: a distinct clinicopathologic entity
associated with the Kaposi's sarcoma-associated herpes virus. Blood.
1996;88:645-56. http://www.ncbi.nlm.nih.gov/pubmed/8695812. PMid:8695812
- Castillo
JJ, Shum H, Lahijani M, Winer ES, Butera JN. Prognosis in primary
effusion lymphoma is associated with the number of body cavities
involved. Leuk Lymphoma. 2012;53:2378-82. https://doi.org/10.3109/10428194.2012.694075
- Gaidano
G, Gloghini A, Gattei V, Rossi MF, Cilia AM, Godeas C, Degan M, Perin
T, Canzonieri V, Aldinucci D, Saglio G, Carbone A, Pinto A. Association
of Kaposi's sarcoma-associated herpesvirus-positive primary effusion
lymphoma with expression of the CD138/syndecan-1 antigen. Blood.
1997;90:4894-900. http://www.ncbi.nlm.nih.gov/pubmed/9389706. PMid:9389706
- Carbone
A, Gloghini A, Larocca LM, Capello D, Pierconti F, Canzonieri V,
Tirelli U, Dalla-Favera R, Gaidano G. Expression profile of MUM1/IRF4,
BCL-6, and CD138/syndecan-1 defines novel histogenetic subsets of human
immunodeficiency virus-related lymphomas. Blood. 2001;97:744-51. https://doi.org/10.1182/blood.V97.3.744 PMid:11157493
- Tsuboi
K, Iida S, Inagaki H, Kato M, Hayami Y, Hanamura I, Miura K, Harada S,
Kikuchi M, Komatsu H, Banno S, Wakita A, Nakamura S, Eimoto T, Ueda R.
MUM1/IRF4 expression as a frequent event in mature lymphoid
malignancies. Leukemia. 2000;14:449-56, https://doi.org/10.1038/sj.leu.2401696 PMid:10720141
- Chadburn
A, Hyjek EM, Tam W, Liu Y, Rengifo T, Cesarman E, Knowles DM.
Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV;
HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD).
Histopathology. 2008;53:513-24. https://doi.org/10.1111/j.1365-2559.2008.03144.x
- Riva
G, Lagreca I, Mattiolo A, Belletti D, Lignitto L, Barozzi P, Ruozi B,
Vallerini D, Quadrelli C, Corradini G, Forghieri F, Marasca R, Narni F,
Tosi G, Forni F, Vandelli MA, Amadori A, Chieco-Bianchi L, Potenza L,
Calabro ML, Luppi M. Antineoplastic effects of liposomal short
interfering RNA treatment targeting BLIMP1/PRDM1 in primary effusion
lymphoma. Haematologica. 2015;100:e467-70. https://doi.org/10.3324/haematol.2015.126854
- Szatmari
T, Otvos R, Hjerpe A, Dobra K. Syndecan-1 in Cancer: Implications for
Cell Signaling, Differentiation, and Prognostication. Dis Markers.
2015;2015:796052. https://doi.org/10.1155/2015/796052
- Wijdenes
J, Vooijs WC, Clement C, Post J, Morard F, Vita N, Laurent P, Sun RX,
Klein B, Dore JM. A plasmocyte selective monoclonal antibody (B-B4)
recognizes syndecan-1. Br J Haematol. 1996;94:318-23, https://doi.org/10.1046/j.1365-2141.1996.d01-1811.x PMid:8759892
- Klein
U, Gloghini A, Gaidano G, Chadburn A, Cesarman E, Dalla-Favera R,
Carbone A. Gene expression profile analysis of AIDS-related primary
effusion lymphoma (PEL) suggests a plasmablastic derivation and
identifies PEL-specific transcripts. Blood. 2003;101:4115-21. https://doi.org/10.1182/blood-2002-10-3090
- Foussat
A, Wijdenes J, Bouchet L, Gaidano G, Neipel F, Balabanian K, Galanaud
P, Couderc J, Emilie D. Human interleukin-6 is in vivo an autocrine
growth factor for human herpesvirus-8-infected malignant B lymphocytes.
Eur Cytokine Netw. 1999;10:501-8, http://www.ncbi.nlm.nih.gov/pubmed/10586116 PMid:10586116
- Drexler
HG, Meyer C, Gaidano G, Carbone A. Constitutive cytokine production by
primary effusion (body cavity-based) lymphoma-derived cell lines.
Leukemia. 1999;13:634-40, https://doi.org/10.1038/sj.leu.2401371 PMid:10214873
- Calabro
ML, Gasperini P, Di Gangi IM, Indraccolo S, Barbierato M, Amadori A,
Chieco-Bianchi L. Antineoplastic activity of lentiviral vectors
expressing interferon-alpha in a preclinical model of primary effusion
lymphoma. Blood. 2009;113:4525-33. https://doi.org/10.1182/blood-2008-09-180307
- Aoki
Y, Tosato G. Role of vascular endothelial growth factor/vascular
permeability factor in the pathogenesis of Kaposi's sarcoma-associated
herpesvirus-infected primary effusion lymphomas. Blood.
1999;94:4247-54. http://www.ncbi.nlm.nih.gov/pubmed/10590069 PMid:10590069
- Gaidano
G, Carbone A. Primary effusion lymphoma: a liquid phase lymphoma of
fluid-filled body cavities. Adv Cancer Res. 2001;80:115-46.
https://doi.org/10.1016/S0065-230X(01)80014-2
- Simonelli
C, Spina M, Cinelli R, Talamini R, Tedeschi R, Gloghini A, Vaccher E,
Carbone A, Tirelli U. Clinical features and outcome of primary effusion
lymphoma in HIV-infected patients: a single-institution study. J Clin
Oncol. 2003;21:3948-54. https://doi.org/10.1200/JCO.2003.06.013
- Boulanger
E, Gerard L, Gabarre J, Molina JM, Rapp C, Abino JF, Cadranel J,
Chevret S, Oksenhendler E. Prognostic factors and outcome of human
herpesvirus 8-associated primary effusion lymphoma in patients with
AIDS. J Clin Oncol. 2005;23:4372-80. https://doi.org/10.1200/JCO.2005.07.084
- Boulanger
E, Daniel MT, Agbalika F, Oksenhendler E. Combined chemotherapy
including high-dose methotrexate in KSHV/HHV8-associated primary
effusion lymphoma. Am J Hematol. 2003;73:143-8. https://doi.org/10.1002/ajh.10341
- Guillet
S, Gerard L, Meignin V, Agbalika F, Cuccini W, Denis B, Katlama C,
Galicier L, Oksenhendler E. Classic and extracavitary primary effusion
lymphoma in 51 HIV-infected patients from a single institution. Am J
Hematol. 2016;91:233-7. https://doi.org/10.1002/ajh.24251
- Ascoli
V, Lo Coco F, Torelli G, Vallisa D, Cavanna L, Bergonzi C, Luppi M.
Human herpesvirus 8-associated primary effusion lymphoma in
HIV--patients: a clinicopidemiologic variant resembling classic
Kaposi's sarcoma. Haematologica. 2002;87:339-43. http://www.ncbi.nlm.nih.gov/pubmed/11940475 PMid:11940475
- Boulanger
E, Hermine O, Fermand JP, Radford-Weiss I, Brousse N, Meignin V,
Gessain A. Human herpesvirus 8 (HHV-8)-associated peritoneal primary
effusion lymphoma (PEL) in two HIV-negative elderly patients. Am J
Hematol. 2004;76:88-91. https://doi.org/10.1002/ajh.20048
- Klepfish
A, Sarid R, Shtalrid M, Shvidel L, Berrebi A, Schattner A. Primary
effusion lymphoma (PEL) in HIV-negative patients--a distinct clinical
entity. Leuk Lymphoma. 2001;41:439-43. https://doi.org/10.3109/10428190109058002
- Riva
G, Luppi M, Barozzi P, Forghieri F, Potenza L. How I treat
HHV8/KSHV-related diseases in posttransplant patients. Blood.
2012;120:4150-9. https://doi.org/10.1182/blood-2012-04-421412
- Boulanger
E, Afonso PV, Yahiaoui Y, Adle-Biassette H, Gabarre J, Agbalika F.
Human herpesvirus-8 (HHV-8)-associated primary effusion lymphoma in two
renal transplant recipients receiving rapamycin. Am J Transplant.
2008;8:707-10. https://doi.org/10.1111/j.1600-6143.2007.02110.x
- Christenson
ES, Teply B, Agrawal V, Illei P, Gurakar A, Kanakry JA. Human
Herpesvirus 8-Related Primary Effusion Lymphoma After Liver
Transplantation. Am J Transplant. 2015;15:2762-6. https://doi.org/10.1111/ajt.13321
- Nakayama-Ichiyama
S, Yokote T, Kobayashi K, Hirata Y, Hiraoka N, Iwaki K, Takayama A,
Akioka T, Oka S, Miyoshi T, Fukui H, Tsuda Y, Takubo T, Tsuji M,
Higuchi K, Hanafusa T. Primary effusion lymphoma of T-cell origin with
t(7;8)(q32;q13) in an HIV-negative patient with HCV-related liver
cirrhosis and hepatocellular carcinoma positive for HHV6 and HHV8. Ann
Hematol. 2011;90:1229-31. https://doi.org/10.1007/s00277-011-1165-8
- Gandhi SA, Mufti G, Devereux S, Ireland R. Primary effusion lymphoma in an HIV-negative man. Br J Haematol. 2011;155:411. https://doi.org/10.1111/j.1365-2141.2011.08778.x
- Ensoli
B, Sturzl M, Monini P. Cytokine-mediated growth promotion of Kaposi's
sarcoma and primary effusion lymphoma. Semin Cancer Biol.
2000;10:367-81. https://doi.org/10.1006/scbi.2000.0329
- Boulanger
E, Duprez R, Delabesse E, Gabarre J, Macintyre E, Gessain A.
Mono/oligoclonal pattern of Kaposi Sarcoma-associated herpesvirus
(KSHV/HHV-8) episomes in primary effusion lymphoma cells. Int J Cancer.
2005;115:511-8. https://doi.org/10.1002/ijc.20926
- Ascoli
V, Calabro ML, Giannakakis K, Barbierato M, Chieco-Bianchi L, Gastaldi
R, Narciso P, Gaidano G, Capello D. Kaposi's sarcoma-associated
herpesvirus/human herpesvirus 8-associated polyclonal body cavity
effusions that mimic primary effusion lymphomas. Int J Cancer.
2006;119:1746-8; author reply 49-50. https://doi.org/10.1002/ijc.21965
- Mutsaers SE. Mesothelial cells: their structure, function and role in serosal repair. Respirology. 2002;7:171-91. https://doi.org/10.1046/j.1440-1843.2002.00404.x PMid:12153683
- Ensoli B, Sturzl M, Monini P. Reactivation and role of HHV-8 in Kaposi's sarcoma initiation. Adv Cancer Res. 2001;81:161-200. https://doi.org/10.1016/S0065-230X(01)81005-8
- Mutsaers SE, Prele CM, Pengelly S, Herrick SE. Mesothelial cells and peritoneal homeostasis. Fertil Steril. 2016;106:1018-24. https://doi.org/10.1016/j.fertnstert.2016.09.005
- Sanchez-Martin
D, Uldrick TS, Kwak H, Ohnuki H, Polizzotto MN, Annunziata CM, Raffeld
M, Wyvill KM, Aleman K, Wang V, Marshall VA, Whitby D, Yarchoan R,
Tosato G. Evidence for a Mesothelial Origin of Body Cavity Effusion
Lymphomas. J Natl Cancer Inst. 2017;109. https://doi.org/10.1093/jnci/djx016
- Lignitto
L, Mattiolo A, Negri E, Persano L, Gianesello L, Chieco-Bianchi L,
Calabro ML. Crosstalk between the mesothelium and lymphomatous cells:
insight into the mechanisms involved in the progression of body cavity
lymphomas. Cancer Med. 2014;3:1-13. https://doi.org/10.1002/cam4.159
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8. https://doi.org/10.1172/JCI39104
- Aroeira
LS, Loureiro J, Gonzalez-Mateo GT, Fernandez-Millara V, del Peso G,
Sanchez-Tomero JA, Ruiz-Ortega M, Bajo MA, Lopez-Cabrera M, Selgas R.
Characterization of epithelial-to-mesenchymal transition of mesothelial
cells in a mouse model of chronic peritoneal exposure to high glucose
dialysate. Perit Dial Int. 2008;28 Suppl 5:S29-33, http://www.ncbi.nlm.nih.gov/pubmed/19008536 PMid:19008536
- Morassut
S, Vaccher E, Balestreri L, Gloghini A, Gaidano G, Volpe R, Tirelli U,
Carbone A. HIV-associated human herpesvirus 8-positive primary
lymphomatous effusions: radiologic findings in six patients. Radiology.
1997;205:459-63. https://doi.org/10.1148/radiology.205.2.9356629
- Chadburn
A, Hyjek E, Mathew S, Cesarman E, Said J, Knowles DM. KSHV-positive
solid lymphomas represent an extra-cavitary variant of primary effusion
lymphoma. Am J Surg Pathol. 2004;28:1401-16, http://www.ncbi.nlm.nih.gov/pubmed/15489644 https://doi.org/10.1097/01.pas.0000138177.10829.5c PMid:15489644
- Boulanger
E, Meignin V, Afonso PV, Duprez R, Oksenhendler E, Agbalika F, Gessain
A. Extracavitary tumor after primary effusion lymphoma: relapse or
second distinct lymphoma? Haematologica. 2007;92:1275-6, https://doi.org/10.3324/haematol.11364 PMid:17768127
- Komanduri
KV, Luce JA, McGrath MS, Herndier BG, Ng VL. The natural history and
molecular heterogeneity of HIV-associated primary malignant
lymphomatous effusions. J Acquir Immune Defic Syndr Hum Retrovirol.
1996;13:215-26, https://doi.org/10.1097/00042560-199611010-00003 PMid:8898666
- Maric
I, Washington S, Schwartz A, Anandan V, Karcher DS. Human
herpesvirus-8-positive body cavity-based lymphoma involving the atria
of the heart: a case report. Cardiovasc Pathol.
2002;11:244-7, https://doi.org/10.1016/S1054-8807(02)00112-6
- Huang
Q, Chang KL, Gaal K, Arber DA. Primary effusion lymphoma with
subsequent development of a small bowel mass in an HIV-seropositive
patient: a case report and literature review. Am J Surg Pathol.
2002;26:1363-7. https://doi.org/10.1097/00000478-200210000-00014 PMid:12360052
- DePond
W, Said JW, Tasaka T, de Vos S, Kahn D, Cesarman E, Knowles DM,
Koeffler HP. Kaposi's sarcoma-associated herpesvirus and human
herpesvirus 8 (KSHV/HHV8)-associated lymphoma of the bowel. Report of
two cases in HIV-positive men with secondary effusion lymphomas. Am J
Surg Pathol. 1997;21:719-24, https://doi.org/10.1097/00000478-199706000-00013 PMid:9199651
- Medeiros
BC, Maness LJ, Bauer FA, Ross JW, Kapur D. Unusual presentation of
"extracavitary" primary effusion lymphoma in previously unknown HIV
disease. Conn Med. 2000;64:591-4, http://www.ncbi.nlm.nih.gov/pubmed/11100630 PMid:11100630
- Dhungel
BM, Montgomery ND, Painschab MS, Mulenga M, Tomoka T, Kaimila B, Zuze
T, Kasonkanji E, Kampani C, Chimzimu F, Randall C, Krysiak R, Seguin R,
Fedoriw Y, Gopal S. 'Discovering' primary effusion lymphoma in Malawi.
AIDS. 2018;32:2264-66. https://doi.org/10.1097/QAD.0000000000001933
- Gessain
A, Briere J, Angelin-Duclos C, Valensi F, Beral HM, Davi F, Nicola MA,
Sudaka A, Fouchard N, Gabarre J, Troussard X, Dulmet E, Audouin J,
Diebold J, de The G. Human herpes virus 8 (Kaposi's sarcoma herpes
virus) and malignant lymphoproliferations in France: a molecular study
of 250 cases including two AIDS-associated body cavity based lymphomas.
Leukemia. 1997;11:266-72.ù https://doi.org/10.1038/sj.leu.2400549 PMid:9009091
- Kim
Y, Leventaki V, Bhaijee F, Jackson CC, Medeiros LJ, Vega F.
Extracavitary/solid variant of primary effusion lymphoma. Ann Diagn
Pathol. 2012;16:441-6. https://doi.org/10.1016/j.anndiagpath.2012.03.004
- Green
I, Espiritu E, Ladanyi M, Chaponda R, Wieczorek R, Gallo L, Feiner H.
Primary lymphomatous effusions in AIDS: a morphological,
immunophenotypic, and molecular study. Mod Pathol. 1995;8:39-45, http://www.ncbi.nlm.nih.gov/pubmed/7731940 PMid:7731940
- Ascoli
V, Sirianni MC, Mezzaroma I, Mastroianni CM, Vullo V, Andreoni M,
Narciso P, Scalzo CC, Nardi F, Pistilli A, Lo Coco F. Human
herpesvirus-8 in lymphomatous and nonlymphomatous body cavity effusions
developing in Kaposi's sarcoma and multicentric Castleman's disease.
Ann Diagn Pathol. 1999;3:357-63. https://doi.org/10.1053/ADPA00300357
- Castleman
B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia
resembling thymoma. Cancer. 1956;9:822-30,
http://www.ncbi.nlm.nih.gov/pubmed/13356266 https://doi.org/10.1002/1097-0142(195607/08)9:4<822::AID-CNCR2820090430>3.0.CO;2-4
- Liu
AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, Krymskaya
VP, Kelleher D, Rubenstein AH, Fajgenbaum DC. Idiopathic multicentric
Castleman's disease: a systematic literature review. Lancet Haematol.
2016;3:e163-75. https://doi.org/10.1016/S2352-3026(16)00006-5
- Chan
KL, Lade S, Prince HM, Harrison SJ. Update and new approaches in the
treatment of Castleman disease. J Blood Med. 2016;7:145-58.
https://doi.org/10.2147/JBM.S60514
- Simpson D. Epidemiology of Castleman Disease. Hematol Oncol Clin North Am. 2018;32:1-10. https://doi.org/10.1016/j.hoc.2017.09.001
- Dupin
N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E,
Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C.
Distribution of human herpesvirus-8 latently infected cells in Kaposi's
sarcoma, multicentric Castleman's disease, and primary effusion
lymphoma. Proc Natl Acad Sci U S A. 1999;96:4546-51, https://doi.org/10.1073/pnas.96.8.4546 PMid:10200299
- Dupin
N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, Weiss RA, Isaacson
PG, Boshoff C. HHV-8 is associated with a plasmablastic variant of
Castleman disease that is linked to HHV-8-positive plasmablastic
lymphoma. Blood. 2000;95:1406-12. http://www.ncbi.nlm.nih.gov/pubmed/10666218 PMid:10666218
- Cobaleda
C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell
identity and function. Nat Immunol. 2007;8:463-70. https://doi.org/10.1038/ni1454
- Du
MQ, Liu H, Diss TC, Ye H, Hamoudi RA, Dupin N, Meignin V, Oksenhendler
E, Boshoff C, Isaacson PG. Kaposi sarcoma-associated herpesvirus
infects monotypic (IgM lambda) but polyclonal naive B cells in
Castleman disease and associated lymphoproliferative disorders. Blood.
2001;97:2130-6. https://doi.org/10.1182/blood.V97.7.2130 PMid:11264181
- Barozzi
P, Luppi M, Masini L, Marasca R, Savarino M, Morselli M, Ferrari MG,
Bevini M, Bonacorsi G, Torelli G. Lymphotropic herpes virus (EBV,
HHV-6, HHV-8) DNA sequences in HIV negative Castleman's disease. Clin
Mol Pathol. 1996;49:M232-5. https://doi.org/10.1136/mp.49.4.M232 PMid:16696081 PMCid:PMC408065
- Corbellino
M, Poirel L, Aubin JT, Paulli M, Magrini U, Bestetti G, Galli M,
Parravicini C. The role of human herpesvirus 8 and Epstein-Barr virus
in the pathogenesis of giant lymph node hyperplasia (Castleman's
disease). Clin Infect Dis. 1996;22:1120-1. https://doi.org/10.1093/clinids/22.6.1120 PMid:8783733
- Gessain
A, Sudaka A, Briere J, Fouchard N, Nicola MA, Rio B, Arborio M,
Troussard X, Audouin J, Diebold J, de The G. Kaposi sarcoma-associated
herpes-like virus (human herpesvirus type 8) DNA sequences in
multicentric Castleman's disease: is there any relevant association in
non-human immunodeficiency virus-infected patients? Blood.
1996;87:414-6. http://www.ncbi.nlm.nih.gov/pubmed/8547672 PMid:8547672
- Chadburn
A, Cesarman E, Nador RG, Liu YF, Knowles DM. Kaposi's
sarcoma-associated herpesvirus sequences in benign lymphoid
proliferations not associated with human immunodeficiency virus.
Cancer. 1997;80:788-97. https://doi.org/10.1002/(SICI)1097-0142(19970815)80:4<788::AID-CNCR18>3.0.CO;2-P
- Fajgenbaum
DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, Simpson D, Liu AY,
Menke D, Chandrakasan S, Lechowicz MJ, Wong RS, Pierson S, Paessler M,
Rossi JF, Ide M, Ruth J, Croglio M, Suarez A, Krymskaya V, Chadburn A,
Colleoni G, Nasta S, Jayanthan R, Nabel CS, Casper C, Dispenzieri A,
Fossa A, Kelleher D, Kurzrock R, Voorhees P, Dogan A, Yoshizaki K, van
Rhee F, Oksenhendler E, Jaffe ES, Elenitoba-Johnson KS, Lim MS.
International, evidence-based consensus diagnostic criteria for
HHV-8-negative/idiopathic multicentric Castleman disease. Blood.
2017;129:1646-57. https://doi.org/10.1182/blood-2016-10-746933
- Polizzotto
MN, Uldrick TS, Hu D, Yarchoan R. Clinical Manifestations of Kaposi
Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease
(KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front
Microbiol. 2012;3:73. https://doi.org/10.3389/fmicb.2012.00073
- Oksenhendler
E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel JP, Agbalika
F. High levels of human herpesvirus 8 viral load, human interleukin-6,
interleukin-10, and C reactive protein correlate with exacerbation of
multicentric castleman disease in HIV-infected patients. Blood.
2000;96:2069-73. http://www.ncbi.nlm.nih.gov/pubmed/10979949 PMid:10979949
- Yoshizaki
K, Matsuda T, Nishimoto N, Kuritani T, Taeho L, Aozasa K, Nakahata T,
Kawai H, Tagoh H, Komori T, et al. Pathogenic significance of
interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood.
1989;74:1360-7. http://www.ncbi.nlm.nih.gov/pubmed/2788466 PMid:2788466
- Brandt
SJ, Bodine DM, Dunbar CE, Nienhuis AW. Dysregulated interleukin 6
expression produces a syndrome resembling Castleman's disease in mice.
J Clin Invest. 1990;86:592-9. https://doi.org/10.1172/JCI114749
- Suthaus
J, Stuhlmann-Laeisz C, Tompkins VS, Rosean TR, Klapper W, Tosato G,
Janz S, Scheller J, Rose-John S. HHV-8-encoded viral IL-6 collaborates
with mouse IL-6 in the development of multicentric Castleman disease in
mice. Blood. 2012;119:5173-81. https://doi.org/10.1182/blood-2011-09-377705
- Debord
C, Eveillard M. Leukemic phase of a large B-cell lymphoma arising in
KSHV-associated multicentric Castleman disease. Blood. 2015;126:1966,
http://www.ncbi.nlm.nih.gov/pubmed/26756053 https://doi.org/10.1182/blood-2015-08-663328 PMid:26756053
- Manogaran C, Kassam S. HHV8-associated lymphoproliferative disorder in the peripheral blood. Blood. 2018;131:2868. https://doi.org/10.1182/blood-2018-04-837823
- Schulte KM, Talat N. Castleman's disease--a two compartment model of HHV8 infection. Nat Rev Clin Oncol. 2010;7:533-43. https://doi.org/10.1038/nrclinonc.2010.103
- Seliem
RM, Griffith RC, Harris NL, Beheshti J, Schiffman FJ, Longtine J, Kutok
J, Ferry JA. HHV-8+, EBV+ multicentric plasmablastic microlymphoma in
an HIV+ Man: the spectrum of HHV-8+ lymphoproliferative disorders
expands. Am J Surg Pathol. 2007;31:1439-45. https://doi.org/10.1097/PAS.0b013e31804d43d8
- Delecluse
HJ, Anagnostopoulos I, Dallenbach F, Hummel M, Marafioti T, Schneider
U, Huhn D, Schmidt-Westhausen A, Reichart PA, Gross U, Stein H.
Plasmablastic lymphomas of the oral cavity: a new entity associated
with the human immunodeficiency virus infection. Blood.
1997;89:1413-20, http://www.ncbi.nlm.nih.gov/pubmed/9028965 PMid:9028965
- Oksenhendler
E, Boulanger E, Galicier L, Du MQ, Dupin N, Diss TC, Hamoudi R, Daniel
MT, Agbalika F, Boshoff C, Clauvel JP, Isaacson PG, Meignin V. High
incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin
lymphoma in patients with HIV infection and multicentric Castleman
disease. Blood. 2002;99:2331-6, https://doi.org/10.1182/blood.V99.7.2331 PMid:11895764
- Castillo
J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma:
lessons learned from 112 published cases. Am J Hematol. 2008;83:804-9. https://doi.org/10.1002/ajh.21250
- Colomo
L, Loong F, Rives S, Pittaluga S, Martinez A, Lopez-Guillermo A,
Ojanguren J, Romagosa V, Jaffe ES, Campo E. Diffuse large B-cell
lymphomas with plasmablastic differentiation represent a heterogeneous
group of disease entities. Am J Surg Pathol. 2004;28:736-47. https://doi.org/10.1097/01.pas.0000126781.87158.e3 PMid:15166665
- Dolcetti R, Gloghini A, Caruso A, Carbone A. A lymphomagenic role for HIV beyond immune suppression? Blood. 2016;127:1403-9. https://doi.org/10.1182/blood-2015-11-681411
- Gonzalez-Farre
B, Martinez D, Lopez-Guerra M, Xipell M, Monclus E, Rovira J, Garcia F,
Lopez-Guillermo A, Colomo L, Campo E, Martinez A. HHV8-related lymphoid
proliferations: a broad spectrum of lesions from reactive lymphoid
hyperplasia to overt lymphoma. Mod Pathol. 2017;30:745-60. https://doi.org/10.1038/modpathol.2016.233
- Polizzotto
MN, Uldrick TS, Wyvill KM, Aleman K, Marshall V, Wang V, Whitby D,
Pittaluga S, Jaffe ES, Millo C, Tosato G, Little RF, Steinberg SM,
Sereti I, Yarchoan R. Clinical Features and Outcomes of Patients With
Symptomatic Kaposi Sarcoma Herpesvirus (KSHV)-associated Inflammation:
Prospective Characterization of KSHV Inflammatory Cytokine Syndrome
(KICS). Clin Infect Dis. 2016;62:730-38. https://doi.org/10.1093/cid/civ996
- Luppi
M, Barozzi P, Schulz TF, Setti G, Staskus K, Trovato R, Narni F,
Donelli A, Maiorana A, Marasca R, Sandrini S, Torelli G. Bone marrow
failure associated with human herpesvirus 8 infection after
transplantation. N Engl J Med. 2000;343:1378-85. https://doi.org/10.1056/NEJM200011093431905
- Pietrosi
G, Vizzini G, Pipitone L, Di Martino G, Minervini MI, Lo Iacono G,
Conaldi PG, Grossi P, Lamonaca V, Galatioto L, Gruttadauria S, Gridelli
B. Primary and reactivated HHV8 infection and disease after liver
transplantation: a prospective study. Am J Transplant. 2011;11:2715-23.
https://doi.org/10.1111/j.1600-6143.2011.03769.x
- Mularoni
A, Gallo A, Riva G, Barozzi P, Miele M, Cardinale G, Vizzini G, Volpes
R, Grossi P, Di Carlo D, Luca A, Trenti T, Luppi M, Conaldi PG.
Successful Treatment of Kaposi Sarcoma-Associated Herpesvirus
Inflammatory Cytokine Syndrome After Kidney-Liver Transplant:
Correlations With the Human Herpesvirus 8 miRNome and Specific T Cell
Response. Am J Transplant. 2017;17:2963-69. https://doi.org/10.1111/ajt.14346
- Bhavsar
T, Lee JC, Perner Y, Raffeld M, Xi L, Pittaluga S, Jaffe ES.
KSHV-associated and EBV-associated Germinotropic Lymphoproliferative
Disorder: New Findings and Review of the Literature. Am J Surg Pathol.
2017;41:795-800. https://doi.org/10.1097/PAS.0000000000000823
- Coupland
SE, Charlotte F, Mansour G, Maloum K, Hummel M, Stein H.
HHV-8-associated T-cell lymphoma in a lymph node with concurrent
peritoneal effusion in an HIV-positive man. Am J Surg Pathol.
2005;29:647-52, http://www.ncbi.nlm.nih.gov/pubmed/15832089 https://doi.org/10.1097/01.pas.0000157937.01624.1d PMid:15832089
- Said
JW, Shintaku IP, Asou H, deVos S, Baker J, Hanson G, Cesarman E, Nador
R, Koeffler HP. Herpesvirus 8 inclusions in primary effusion lymphoma:
report of a unique case with T-cell phenotype. Arch Pathol Lab Med.
1999;123:257-60. https://doi.org/10.1043/0003-9985(1999)123<0257:HIIPEL>2.0.CO;2
- Said
JW, Tasaka T, Takeuchi S, Asou H, de Vos S, Cesarman E, Knowles DM,
Koeffler HP. Primary effusion lymphoma in women: report of two cases of
Kaposi's sarcoma herpes virus-associated effusion-based lymphoma in
human immunodeficiency virus-negative women. Blood. 1996;88:3124-8, http://www.ncbi.nlm.nih.gov/pubmed/8874212 PMid:8874212
- Moatamed
NA, Song SX, Apple SK, Said JW. Primary effusion lymphoma involving the
cerebrospinal fluid. Diagn Cytopathol. 2012;40:635-8. https://doi.org/10.1002/dc.21653
- Courville
EL, Sohani AR, Hasserjian RP, Zukerberg LR, Harris NL, Ferry JA.
Diverse clinicopathologic features in human herpesvirus 8-associated
lymphomas lead to diagnostic problems. Am J Clin Pathol.
2014;142:816-29. https://doi.org/10.1309/AJCPULI3W6WUGGPY
[TOP]