Naouel Guirat Dhouib1, Monia Ben Khaled1, Monia Ouederni1, Imen Ben-Mustapha2, Ridha Kouki1, Habib Besbes1, Mohamed Ridha Barbouche2, Fethi Mellouli1 and Mohamed Bejaoui1.
1 Pediatric Immunohematology Department, Bone Marrow Transplantation Center Tunis, Tunisia.
2
Laboratory of Transmission, Control and Immunobiology of Infections
(LR11IPT02), Institut Pasteur de Tunis, 1002 Tunis, Belvédère, Tunisia.
Correspondence to: Naouel Guirat Dhouib, Pediatric Immunohematology
Department, Bone Marrow Transplantation Center Tunis, Tunisia. Tel: +
216 98 644165. E-mail:
nawel.guirat@yahoo.fr
Published: November 1, 2018
Received: August 27, 2018
Accepted: October 15, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018065 DOI
10.4084/MJHID.2018.065
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Skin
manifestations are frequent among patients with primary
immunodeficiency diseases (PIDs). Their prevalence varies according to
the type of immunodeficiency. This review provides the reader with an
up-to-date summary of the common dermatologic manifestations of PIDs
among Tunisian children. We conducted a prospective study on two
hundred and ninety children with immune deficiency. Demographic details
(including age, sex, and consanguinity) with personal and family
history were recorded. Special attention was paid to cutaneous
manifestations. Dermatological involvements were grouped according to
the etiology of their most prominent sign. Cutaneous manifestations
were found in 164 patients (56.5%). They revealed the diagnosis of PIDs
in 71 patients (24.5 %). The mean age at presentation was 21 months.
Overall the most prominent cutaneous alterations were infectious. They
accounted for 106 cases (36.55%). The most prevalent causes of
cutaneous infections were bacterial: 93 cases (32.06%). Immuno-allergic
skin diseases were among the common findings in our study. These
include eczematous dermatitis found in 62 cases (21.38%). Malignancy
related PIDs was seen in a boy with Wiskott Aldrich syndrome. He
developed Kaposi’s sarcoma at the age of 14 months. Cutaneous changes
are common among children with PIDs. In pediatric patients with failure
to thrive, chronic refractory systemic manifestations often present in
other family members, recurrent cutaneous infections unresponsive to
adequate therapy, atypical forms of eczematous dermatitis or unusual
features should arouse the suspicion of PIDs and prompt specialized
immunologic consultation should be made.
|
Introduction
Primary
immunodeficiency disorders (PIDs) refer to a heterogeneous group of
rare disorders characterized by poor or absent function of one or more
components of the immune system. Over 270 different disorders have been
identified to date, with new disorders continually being recognized.[1]
The clinical presentation of PIDs is highly variable; however, most
disorders involve increased susceptibility to infection. Cutaneous
manifestations are common in PIDs, affecting half of the pediatric
cases and often precede the final diagnosis. Skin infections
characterize many primary immune deficiencies, but noninfectious
cutaneous involvements are frequent including allergic, inflammatory,
autoimmune and malignant manifestations.[2] Only few
studies describing the spectrum of skin disorders in PID are available,
and no similar studies have been conducted in Tunisia. This prospective
study provides the reader with an up-to-date summary of the common
dermatologic manifestations of primary immune deficiency diseases among
Tunisian children.
Materials and Methods
We
conducted a prospective study on two hundred and ninety children
referrals children with suspected immune deficiency during a 10-year
period (January 1, 2008, to December 31, 2017) at the Pediatric
Immunohematology Department in the National Bone Marrow Transplantation
Center in Tunis. Individuals with human immunodeficiency virus
infection or receiving immunosuppressive therapy were excluded from the
study. Demographic details (including age, sex, and consanguinity) with
personal and family history were recorded. Special attention was paid
to cutaneous manifestations. Cutaneous manifestations of PIDs were
classified into four categories: cutaneous lesions of infectious
origin, immunoallergic/autoimmune skin manifestations, pathognomonic
findings related to complex phenotypes and skin cancers associated with
PIDs. The diagnosis of PIDs was established according to the criteria
defined by the International Union of Immunological Societies Expert
Committee for Primary Immunodeficiency.[1] Informed written consent to publish images was obtained from parents.
Results
Among the two hundred and ninety children (Table 1),
111 (38%) were female, and 179 (62%) were male. Consanguineous
relationships were seen in almost two-thirds of patients (65.17%).
Cutaneous manifestations were found in 164 patients (56.5%). They
revealed the diagnosis of PIDs in 71 patients (24.5 %). The mean age at
presentation was 21 months (1 day-25 years). The most predominant
cutaneous alterations were infectious in 106 cases (36.55%). The most
prevalent causes of cutaneous infections were bacterial: 93 cases,
(32.06%).
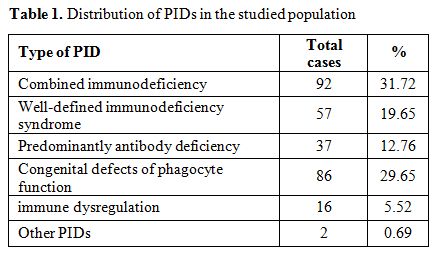 |
Table 1.
Distribution of PIDs in the studied population |
Staphylococcus aureus represented by far the most common pathogen (32 cases), and Pseudomonas
was responsible for one other case. Unfortunately bacterial culture was
not done in all infections. BCG infections were common among patients
with combined immunodeficiency (CID) and septic granulomatous disease
while anal margin abscesses were frequently seen in phagocytic
disorders (Table 2). The next
type of skin infection consisted of viral infections seen in 11 (3.8%)
of the studied patients. Most of them had warts. Recurrent oral
papillomatous lesions related to human papillomavirus were seen in 2
sisters with major histocompatibility complex class II deficiency (Figure 1). The third type was fungal skin infection (Table 2).
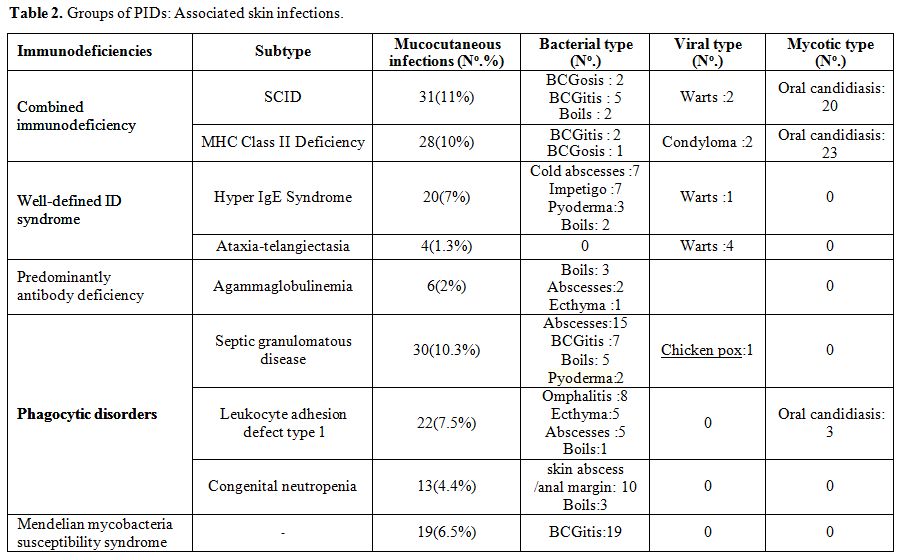 |
Table 2. Groups of PIDs: Associated skin infections. |
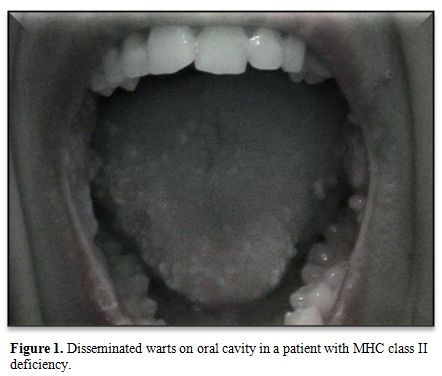 |
Figure 1.
Disseminated warts on oral cavity in a patient with MHC class II deficiency. |
In
this study, oral candidiasis was the most common infection in patients
with CID, frequently associated with broncho-pulmonary infections (61
cases, 66.30%), digestive infections (56 cases, 60.9%), ear, nose, and
throat infections (23 cases, 25%) and persistent diarrhea resulting in
failure to thrive (39 cases, 42.4%). Mucocutaneous lesions due to
candida were more frequently oral, whitish, adherent plaques and
paronychia (Figure 2).
Immuno-allergic skin diseases were among the common findings in our
study. These include eczematous dermatitis found in 62 cases (21.38%), (Table 3). Severely generalized erythroderma with alopecia revealed Omenn syndrome in 17 cases (Figure 3 and 4).
Mean patient age at the onset of these cutaneous manifestations was 6.4
months (1 day to 48 months). Other extracutaneous manifestations led to
the diagnosis including recurrent infections observed in 11 patients
with failure to thrive, as well as infiltration of lymphoid organs with
hepatosplenomegaly. Lymphopenia was found in 15 patients
(88 %) and eosinophilia in 16 cases (94%). Specific
muco-cutaneous manifestations of PIDs were seen in 22 cases (7.6%)
among them telangiectasia (Figure 5)
revealed ataxia telangiectasia syndrome in 14 cases (100%). Among these
patients, three had hypopigmented and café-au-lait macules. Cutaneous
albinism was noted in 8 other cases with Chediack Higashi syndrome
(five cases), Griscelli syndrome (one case) or Hermanski pudlak type 2
syndrome (two cases). Malignancy related PIDs was seen in a boy with
Wiskott Aldrich syndrome. He developed Kaposi’s sarcoma at the age of
14 months (Figure 6).
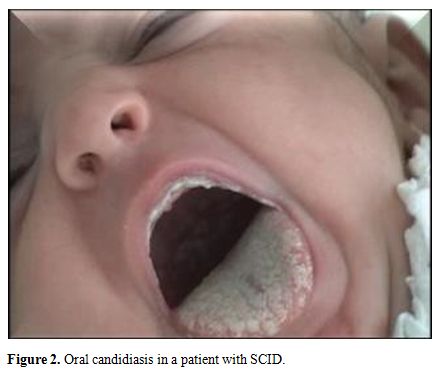 |
Figure 2.
Oral candidiasis in a patient with SCID. |
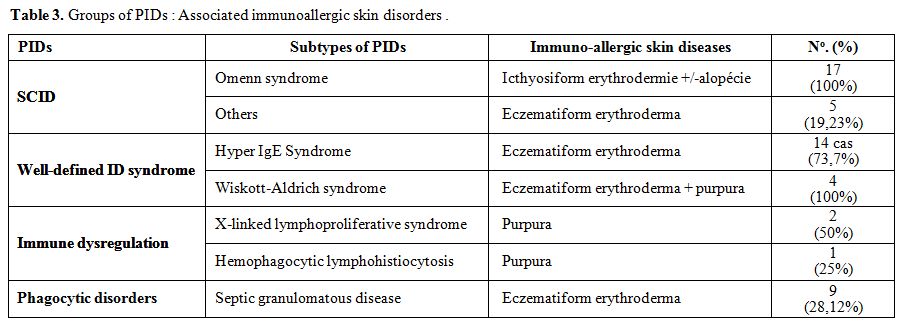 |
Table 3. Groups of PIDs: Associated immunoallergic skin disorders. |
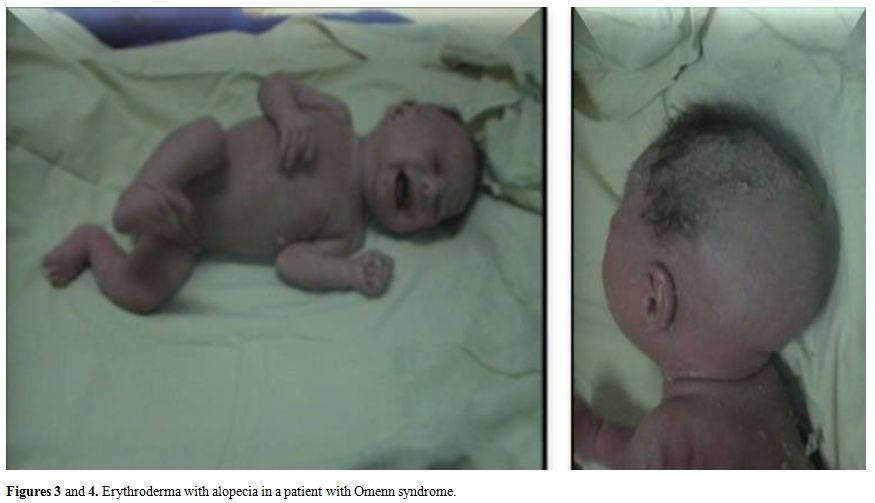 |
Figures 3 and 4. Erythroderma with alopecia in a patient with Omenn syndrome.. |
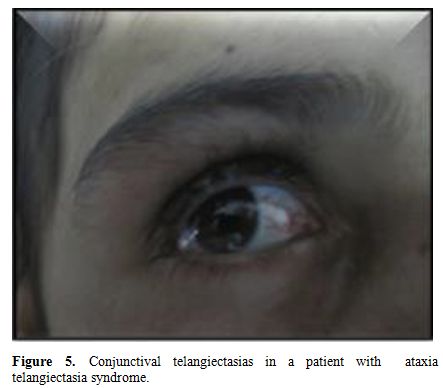 |
Figure 5. Conjunctival telangiectasias in a patient with ataxia telangiectasia syndrome. |
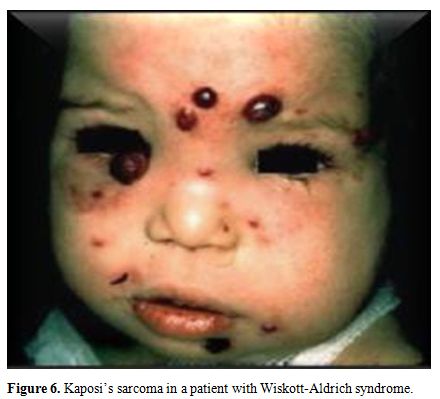 |
Figure 6. Kaposi’s sarcoma in a patient with Wiskott-Aldrich syndrome. |
Discussion
Cutaneous
manifestations may be the clue for the diagnosis of PIDs because of
their early appearance, their high frequency and their easy access for
examination.[3] This study showed that cutaneous
alterations were the presenting problem in 56.5% of cases, which are
higher than what is reported by Moin et al., (32%) and Al-Herz et al.,
(48%).[4,5] Although skin infections were the
most common manifestations, the prevalence rate of infections in our
series was lower than those reported in other countries.[4,6]
Pyogenic infections of the skin frequently occur in PIDs causing
abscesses, boils, ecthyma, cellulitis, folliculitis, or impetigo (Table 2). In this study, recurrent abscesses were frequent. They predominated in phagocytic defects and hyper IgE syndrome.[7]
They tended to be multiple, recurrent and difficult to control.
Bacteriologic cultures should be performed since the growth of unusual
organisms is not infrequent. The most common organism is Staphylococcus followed by Haemophilus, Serratia, Klebsiella, Escherichia coli, and Pseudomonas. In leucocyte adhesion deficiency, recurrent skin infections, generally due to Staphylococcus aureus
or Gram negative bacilli is typical. It tends to necrotize and
ulcerate without pus at the wound site. Other features include
delayed umbilical cord separation, omphalitis, persistent leukocytosis,
and severe destructive gingivitis and periodontitis leading to tooth
loss and alveolar bone resorption.[8] Mycobacterium
bovis in Bacille Calmette-Guérin (BCG) vaccine can cause serious
complications in patients with PIDs ranging from prolonged disease with
lymphadenopathy (BCGitis) to disseminated disease (BCGosis).[9]
These complications have been diagnosed in immunocompromised patients
including combined immunodeficiency, mendelian susceptibility to
mycobacterial disease, chronic granulomatous disease, complete DiGeorge
syndrome, hyper-IgM and hyper-IgE syndromes.[10,11]
The median age of onset is three months of age. Our study showed that
severe combined immunodeficiencies (SCID), MHC Class II Deficiency, CGD
and mendelian susceptibility to mycobacterial disease are the most
common forms of immunodeficiencies among children with BCGosis/BCGitis.
A large study conducted by De Beaucoudrey et al., reported 108 cases
with IL12R β1 deficiency, among them 84 cases presented BCGitis.[12] Similar results were reported by Galal et al.[10]
In our study, 10 out of 13 patients with mendelian susceptibility to
mycobacterial disease had developed suppurative BCG lymphadenitis.
Since the routine national vaccination program in Tunisia includes BCG
vaccine, PIDs, should be considered in any patient with prolonged
BCGitis or BCGosis. Awareness of this phenomenon is important because
these patients need to be treated with antitubercular drugs. Children
with PID are rarely exposed to common viral skin infections as viral
wart and molluscum contagiosum contrasting, thus, with their high
frequency in patients with acquired immunodeficiency syndrome. Herpes
simplex virus is among the most common viral skin infections
encountered in T-cell deficient patients. The symptoms are often
impressive and are relatively resistant to conventional treatment.[4,5]
In the present study, recurrent oral papillomatous lesions related to
human papillomavirus were seen in two sisters with major
histocompatibility complex class II deficiency.[13]
Warts, described as benign skin growths that develop on different parts
of the body and can take on various forms, disseminated warts were
found in 2 patients with WHIM syndrome and ataxia telangiectasia.
Persistent mucocutaneous candidiasis may
be the initial presenting sign for PID in infancy, especially
in those with severe combined immunodeficiency disease.[2,5]
In this study, 76.92% (20/26 cases) with severe combined
immunodeficiency had at least one episode of oral candidiasis.
Candidiasis associated with eczema is a common feature of Hyper IgE
syndrome.[2] When present with mucocutaneous
candidiasis, autoimmune endocrinopathies, and dystrophy of the dental
enamel and nails, it is important to recall the autoimmune
polyendocrinopathy- candidiasis-ectodermal dystrophy syndrome. This
syndrome is caused by mutations in the autoimmune regulator gene
(AIRE), resulting in autoreactive T-cells which develop autoantibodies.[14]
Immunodeficiency should be suspected when skin infection have a
multifocal character, saprophytic or opportunistic and/or a combination
of several pathogens as well as their persistence or their repetition
despite a good anti- infective treatment or when there are skin changes
leading to diagnosis difficulties. Eczematous dermatitis is another
nonspecific cutaneous finding among several PIDs including IgA
deficiency, Immune dysregulation, polyendocrinopathy and enteropathy,
common variable immunodeficiency, Wiskott-Aldrich
syndrome, X-linked agammaglobulinemia, hyper IgM syndromes, hyper
IgE, Netherton’s syndrome and Omenn syndrome. The overall prevalence of
eczema in our patient population (21.38%) was comparable with that in
Mexico (22%) but higher than that reported from Libya (12%).[6,15]
Although
widely present in PIDs, eczema was a consistent feature in patients
with Wiskott- Aldrich syndrome(WAS), a complex and severe X-linked
disorder characterized by microthrombocytopenia, eczema, increased
susceptibility to infection, and increased risk in developing
autoimmunity and lymphomas.[16] Other cutaneous and
mucosal manifestations and hemorrhages are frequent in WAS patients
ranging from non-life-threatening (epistaxis, petechiae, purpura, oral
bleeding) to severe manifestations. This heterogeneous syndrome may be
complicated by autoimmune mucocutaneous manifestations including
Henoch–Schönlein-like purpura, dermatomyositis and recurrent
angioedema.[17] In a clinical series conducted by
Ouederni, eczema was observed in four patients among 35 MHC Class
II Deficiency patients.[18] An underlying primary
immunodeficiency should also be suspected when a patient shows severe
early-onset, atypical, eczematous dermatitis, refractory to therapy
with a tendency to become extensive and flaring up with systemic
infections chronic diarrhea and failure to thrive[19]
or family history suggestive of immunodeficiency in patients with
severe atopic dermatitis. These features underscore the importance of
eliciting a history of recurrent infections or family history
suggestive of immunodeficiency in patients with severe atopic
dermatitis.[20]
A cutaneous granuloma is a
histopathological diagnosis on a tissue that is usually taken for the
evaluation of the cause of an unexplained nodular swelling in the skin.
Aghamohammadi reported a 27-year-old patient with CVID who presented
with multiple skin granulomas on both hands. The patient had been well
until the age of 20 years when she developed these skin lesions with
frequent upper respiratory infections and recurrent diarrhea.
Intravenous immunoglobulin therapy improved skin lesions. In our study,
one patient with CVID developed annular granuloma of the right leg.
Erythroderma
with diffuse alopecia was the third most common skin manifestation in
children with SCID and a consistent presenting feature in infants with
Omenn syndrome, a rare inherited combined immunodeficiency caused by
mutations in various SCID genes. Erythroderma can occur from birth or
develop during the first weeks of life. It is evocative by its
infiltrated character. These patients show hypereosinophilia,
associated with T cell infiltration of gut, liver, spleen, and skin
leading to erythroderma, diarrhea, hepatosplenomegaly, alopecia,
recurrent infection, and failure to thrive.
Our findings support the results of other studies that most PIDs have cutaneous features.
These
features, being their aspects typical, are highly suggestive for the
diagnosis of PIDs. One of the specific cutaneous manifestations of PIDs
is telangiectasia. This PID is caused by alterations in the ATM gene
leading to telangiectasia, immunodeficiency with progressive ataxia and
oculomotor apraxia often accompanied by extrapyramidal movement
disorders.[21,22] In most cases, telangiectasias
first appear when the child reaches three to five years of age.
Conjunctival telangiectasias are first noted in the interpalpebral
bulbar conjunctiva away from the limbus. Cutaneous telangiectasias are
seen on the ears, palate, bridge of the nose and later extend to the
neck, the dorsum of the hands and feet. In our study all patients with
ataxia telangiectasia had telangiectasia. Other skin manifestations
were reported including abnormalities of pigmentation (hypopigmented
and café-au-lait macules), poikiloderma, seborrheic dermatitis, and
less common findings including acanthosis nigricans and hirsutism.[23]
Hypopigmented macules were found in 2 other patients mainly on the
face, trunk, and hands. In our study, hypopigmented and café- au-lait
macules were seen in three cases. Another specific feature of PIDs is
partial albinism with silvery gray hair. This association is an
evocative or even pathognomonic cutaneous sign of complex PIDs
including Chediak Higashi, Griscelli, Hermansky-Pudlak, and
MAPBP-interacting protein deficiency syndromes.[24]
An association of PIDs and cancers has been known for many years[25]
and confirmed from data collected in established registries. The
overall risk for cancer developing in children with PIDs is estimated
to range from 4 to 25%.[26] In a recent study from
the United States Immune Deficiency Network conducted among 3658
patients, 171 separate cancers were diagnosed 25 (15%) of them were
skin cancers. There were 119 cancers (70%) in subjects with CVID.
Thirteen cancers (9%) were observed in subjects with
hypogammaglobulinema and agammaglobulinemia, and eight cancers
(4.6%) were observed in subjects with WAS.[27] Despite
the relatively high number of chronic granulomatous disease patients in
the registry (483), none were diagnosed with cancer. One of the studied
patients with Wiskott Aldrich syndrome developed a Kaposi’s sarcoma
at the age of 14 months.To our
knowledge, this patient is the first case described in the
literature.[28]
Conclusions
Cutaneous
changes may be the presenting signs of PIDs and serve as important
clues for pediatric patients with PIDs. They may also be life-saving in
patients with severe PIDs. Early diagnosis and treatment will prevent
associated morbidity and mortality and improve the quality of life. In
pediatric patients with failure to thrive, chronic refractory
systemic manifestations often present in other family members,
recurrent cutaneous infections unresponsive to adequate therapy,
atypical forms of eczematous dermatitis or unusual features should
arouse the suspicion of PIDs and prompt specialized immunologic
consultation should be made.
References
- Picard C, Al-Herz W, Bousfiha A, Casanova JL,
Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Holland SM,
Klein C, Nonoyama S, Ochs HD, Oksenhendler E, Puck JM, Sullivan KE,
Tang ML, Franco JL, Gaspar HB. Primary Immunodeficiency Diseases: an
Update on the Classification from the International Union of
Immunological Societies Expert Committee for Primary Immunodeficiency
2015. J Clin Immunol. 2015 Nov; 35(8):696-726 http://dx.doi.org/10.1007/s10875-015-0201-1 PMID:26482257
- Lehman H. Skin manifestations of primary immune deficiency. Clin Rev Allergy Immunol 2014;46:112 119 http://dx.doi.org/10.1007/s12016-013-8377-8 PMID:23760761
- Smitt
JHS, Wulffraat NM, Kuijpers TW. The skin in primary immunodeficiency
disorders. Eur J Dermatol 2005;15: 425- 432. PMID:16280293
- Moin
A , Farhoudi A, Moin M, Pourpak Z, Bazargan N. Cutaneous Manifestations
of Primary Immunodeficiency Diseases in Children. Iran J Allergy Asthma
Immunol 2006;5:121-126. http://dx.doi.org/05.03/ijaai.121126 PMID:17237563
- Al Herz W, Nanda A. Skin manifestations in primary immunodeficient children. Pediatr Dermatol 2011; 28:494 501. http://dx.doi.org/10.1111/j.1525-1470.2011.01409.x PMID:21453308
- Suleman
Elfaituri S, Matoug I .Cutaneous Manifestations of Primary
Immunodeficiency Diseases in Libyan Children. J Clin Dermatol Ther
2017;4: 025. http://dx.doi.org/10.24966/CDT-8771/100025
- Yu
JE, Azar AE, Chong HJ, Jongco AM 3rd, Prince BT. Considerations in the
Diagnosis of Chronic Granulomatous Disease. J Pediatric Infect Dis Soc
2018; 7(suppl 1):S6-S11. http://dx.doi.org/10.1093/jpids/piy007 PMID:29746674
- Johnston
S L. Clinical Immunology Review Series: An approach to the patient with
recurrent superficial abscesses. Clin Exp Immunol 2008; 152(3):397–405.
http://dx.doi.org/10.1111/j.1365-2249.2008.03640.x PMID:18422735
- Movahedi
Z, Norouzi S, Mamishi S, Rezaei N. BCGiosis as a presenting feature of
a child with chronic granulomatous disease. Braz J Infect Dis 2011;15:
83-86.PMID:21412596
- Galal N , Boutros J,
Marsafy A, Kong XF, Feinberg J, Casanova JL, Boisson-Dupuis S,
Bustamante J. Mendelian susceptibility to mycobacterial disease in
egyptian children. Mediterr J Hematol Infect Dis 2012. http://dx.doi.org/10.4084/MJHID.2012.033 PMID:22708048
- Norouzi
S, Aghamohammadi A, Mamishi S, et al. Bacillus Calmette-Guérin (BCG)
complications associated with primary immunodeficiency diseases. J
Infect 2012;64:543–54. http://dx.doi.org/10.1016/j.jinf.2012.03.012 PMID:22430715
- De
Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, et
al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from
30 countries. Medicine (Baltimore) 2010; 89: 381-402. http://dx.doi.org/10.1097/MD.0b013e3181fdd832 PMID:21057261
- Guirat-Dhouib
N , Baccar Y, Mustapha IB, Ouederni M, Chouaibi S, El Fekih N,
Barbouche MR, Fezaa B, Kouki R, Hmida S, Mellouli F, Bejaoui M. Oral
HPV infection and MHC class II deficiency (A study of two cases
with atypical outcome). Clin Mol Allergy. 2012 Apr 23;10(1):6. http://dx.doi.org/10.1186/1476-7961-10-6 PMID:22524894
- Collins
SM, Dominguez M, Ilmarinen T, Costigan C, Irvine D. Dermatological
manifestations of autoimmune polyendocrinopathy candidiasis ectodermal
dystrophy syndrome. Br J Dermatol 2006;154:1088 - 1093. http://dx.doi.org/10.1111/j.1365-2133.2006.07166.x PMID:16704638
- Berron-Ruiz
A, Berron-Perez R, Ruiz-Maldonado R. Cutaneous markers of primary
immunodeficiency diseases in children. Pediatr Dermatol. 2000
Mar-;17(2):91-6. PMID:10792794
- Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood 2004;103:456-464. http://dx.doi.org/10.1182/blood-2003-05-1480 PMID:12969986
- Catucci
M, Castiello MC, Pala F, Bosticardo M, Villa A. Autoimmunity in
wiskott-Aldrich syndrome: an unsolved enigma. Front
Immunol.2012;3:209. http://dx.doi.org/10.3389/fimmu.2012.00209 PMID:2282671
- Ouederni
M, Vincent QB, Frange P, et al: Major histocompatibility complex class
II expression deficiency caused by a RFXANK founder mutation: a survey
of 35 patients Blood 2011;118(19):5108-5118. http://dx.doi.org/10.1182/blood-2011-05-352716 PMID:21908431
- Szczawinska-Poplonyk
A, Kycler Z, Pietrucha B, Heropolitanska-Pliszka E, Breborowicz A, et
al. The hyperimmunoglobulin E syndrome - clinical manifestation
diversity in primary immune deficiency. J Rare Dis 2011 ;6:76. http://dx.doi.org/10.1186/1750-1172-6-76 PMID:22085750
- Aghamohammadi
A, Moghaddam ZG, Abolhassani H, Hallaji Z, Mortazavi H, Pourhamdi S, et
al. Investigation of underlying primary immunodeficiencies in patients
with severe atopic dermatitis. Allergol Immunopathol (Madr) 2014;
42(4):336- 341. http://dx.doi.org/10.1016/j.aller.2013.02.004 PMID:23735167
- Chiam
LY, Verhagen MM, Haraldsson A, Wulffraat N, Driessen GJ, Netea MG,
Weemaes CM, Seyger MM, van Deuren M. Cutaneous granulomas in ataxia
telangiectasia and other primary immunodeficiencies: Reflection of
inappropriate immune regulation? Dermatology 2011;223(1):13-19. http://dx.doi.org/10.1159/000330335 PMID:21876338
- Carranza
D, Vega AK, Torres Rusillo S, Montero E, Martinez LJ, Santamaría M, et
al. Molecular and functional characterization of a cohort of Spanish
patients with ataxia telangiectasia. Neuromolecular Med 2017;
19(1):161-174. http://dx.doi.org/10.1007/s12017-016-8440 PMID:27664052
- Greenberger
S , Berkun Y, Ben-Zeev B, Levi YB, Barziliai A, Nissenkorn A.
Dermatologic manifestations of ataxia- telangiectasia syndrome. J Am
Acad Dermatol 2013;68(6):932-6. http://dx.doi.org/10.1016/j.jaad.2012.12.950 PMID:23360865
- Dotta
L, Parolini S, Prandini A, Tabellini G, Antolini M, Kingsmore SF,
Badolato R . Clinical, laboratory and molecular signs of
immunodeficiency in patients with partial oculo- cutaneous albinism.
Orphanet J Rare Dis 2013; 17(8):168. http://dx.doi.org/10.1186/1750-1172-8-168 PMID:24134793
- Notarangelo LD .PIDs and cancer: an evolving story. Blood 2010;116(8):1189-1190. http://dx.doi.org/10.1182/blood-2010-06-286179 PMID:20798238
- Mortaz
E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, Adcock IM.
Cancers Related to Immunodeficiencies: Update and Perspectives. Front
Immunol 2016;7:365.e Collection 2016. http://dx.doi.org/10.3389/fimmu.2016.00365 PMID: 27703456
- Mayor
PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, Fuleihan R,
Garabedian E , Lugar P, Ochs HD, Bonilla FA, Buckley RH, Sullivan KE,
Ballas ZK, Cunningham-Rundles C, Segal BH. Cancer in primary
immunodeficiency diseases: Cancer incidence in the United States Immune
Deficiency Network Registry. J Allergy Clin Immunol.
2018;141(3):1028-1035. http://dx.doi.org/10.1016/j.jaci.2017.05.024 PMID:28606585
- Picard
C , Mellouli F, Duprez R, Chédeville G, Neven B, Fraitag S, Delaunay J,
Le Deist F, Fischer A, Blanche S, Bodemer C, Gessain A, Casanova JL,
Bejaoui M. Kaposi's sarcoma in a child with Wiskott-Aldrich syndrome.
Eur J Pediatr.2006;165(7):453-457. http://dx.doi.org/10.1007/s00431-006-0107-2 PMID:16602009
[TOP]