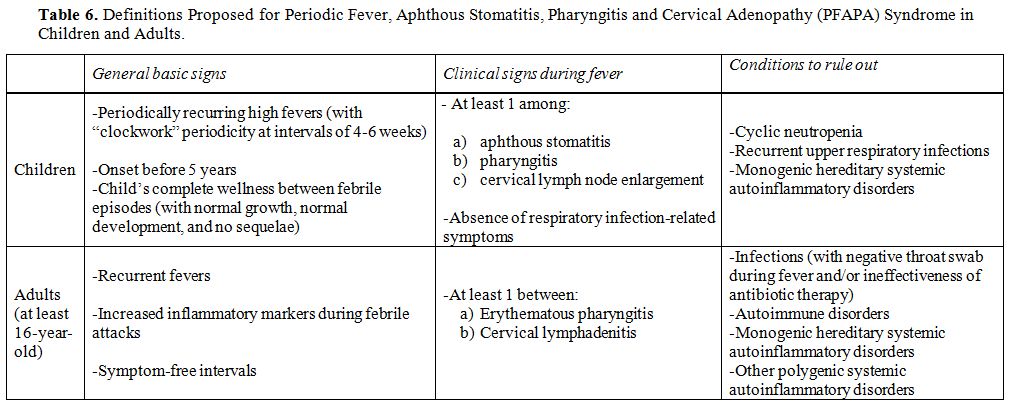
In addition to being crucial for pathogen recognition through the mammalian cysteine protease caspase-1, a central enzyme of innate immunity, which processes pro-interleukin-1 (IL- 1)β into the mature proinflammatory cytokine IL- 1β, the discovery of monogenic and polygenic disorders in which inflammasome activity and IL- 1 release are deregulated highlights the essential importance of inflammasome-supervised human defences against infections.[1] Different studies have demonstrated the existence of a functional hierarchy of proinflammatory cytokines in systemic autoinflammatory disorders (SAIDs), a heterogeneous cluster of rare genetically determined diseases involving innate immunity, mainly characterized by the recurrence of inflammatory flares affecting the skin, joints, serosal membranes, gastroenteric tube, central nervous system and other tissues, in which IL-1β is the most critical driver of inflammation at different levels.[2,3] The term “autoinflammatory” describes the nearly spontaneous appearance of inflammation in the almost complete absence of autoreactive T lymphocytes and/or specific autoantibodies and no clear evidence of infectious triggers.[4] All SAIDs are caused by impaired regulation in the production of proinflammatory cytokines, with IL-1β being the foremost implicated, which leads to a pathological delay in the shutdown of inflammatory responses.[5] The group of SAIDs can be categorized in hereditary monogenic disorders and multifactorial polygenic disorders, encompassing an increasing number of conditions, such as periodic fever, aphthous stomatitis, pharyngitis and cervical adenopathy (PFAPA) syndrome, which is the most relevant cause of idiopathic recurrent fevers in childhood.[6]
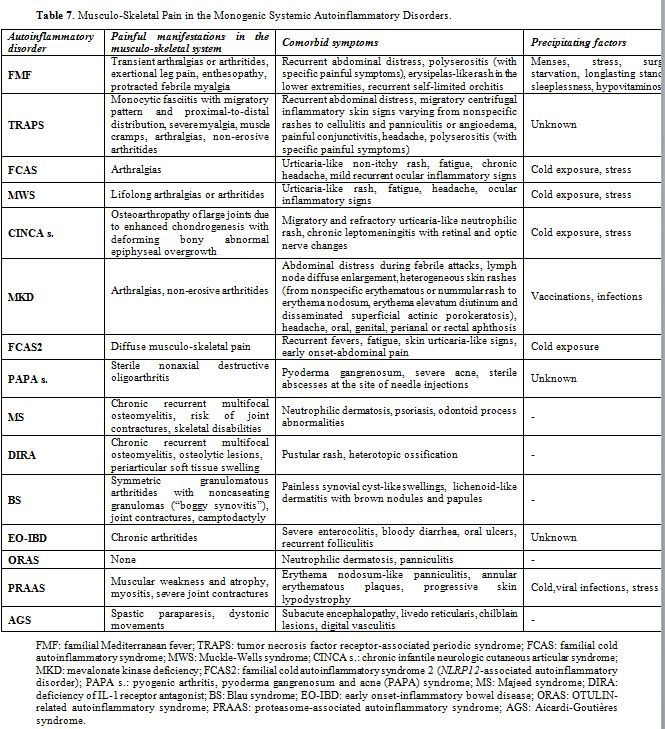
From the historical understanding that monogenic periodic fevers are the prototype of pure SAIDs, our knowledge has now expanded to encompass a larger number of inflammatory diseases with autoinflammatory mechanisms and a presumed polygenic basis, such as Behçet’s disease, gout and idiopathic recurrent acute pericarditis. Further advances in genetics and molecular biology have also consented to classify the hereditary monogenic SAIDs more rationally, though they are characterized by quite similar clinical phenotypes during any flare with fever and varying signs of systemic and sometimes organ- specific inflammation. The group of SAIDs includes familial Mediterranean fever (FMF), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), the family of cryopyrin- associated periodic syndrome (CAPS), which in turn encompasses familial cold urticaria syndrome (FCAS), Muckle-Wells syndrome (MWS) and chronic infantile neurological cutaneous and articular (CINCA) syndrome, mevalonate kinase deficiency (MKD), hereditary pyogenic diseases including pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome, Majeed syndrome (MS) and deficiency of the IL-1 receptor antagonist (DIRA), in addition to idiopathic granulomatous diseases with familiar presentation, such as Blau syndrome (BS), dysregulations in interferon (IFN) signaling and defects of the ubiquitin-proteasome pathway, such as OTULIN-related autoinflammatory syndrome (ORAS) and proteasome-associated autoinflammatory syndrome (PRAAS).
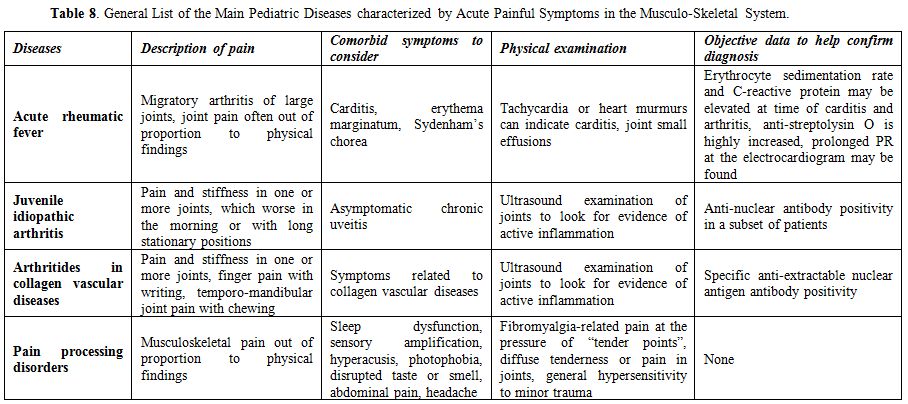
A descriptive summary of the monogenic SAIDs described in this review is listed in the Table 1. Some of these - namely FMF, MKD, MS, DIRA, ORAS, PRAAS - are transmitted by autosomal recessive inheritance, while the others - TRAPS, FCAS, MWS, CINCA syndrome, PAPA syndrome and BS - are autosomal dominant. The genes associated with these diseases have been sequentially identified from 1997 onwards and, with the exception of MKD, the majority of them encodes for proteins involved in the inflammasome activity or in the process of programmed cell death.[7] In particular, inflammasomes, which have a characteristic structure consisting of a central scaffold and sensor protein (a Nod-like receptor or IFN- inducible protein), an adaptor protein ASC (an apoptosis-associated speck-like protein containing a CARD domain) and the effector protein caspase- 1, modulate both synthesis and release of IL-1β; they work - under numerous different activation modes - as innate immunologic platforms assembled in the cytosol in response to the sensing of pathogens or cellular damage.[8]
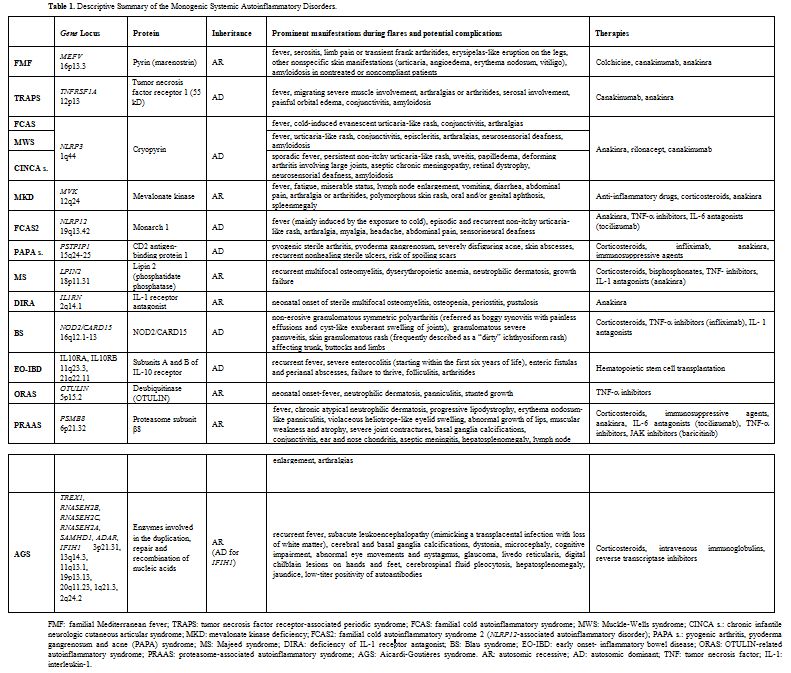 |
Table 1. Descriptive Summary of the Monogenic Systemic Autoinflammatory Disorders. |