Alessandro Malara1-2, Vittorio Abbonante1-2, Maria Zingariello3, Annarita Migliaccio4 and Alessandra Balduini1,2,5.
1 Department of Molecular Medicine, University of Pavia, Pavia, Italy.
2 Biotechnology Research Laboratories, IRCCS San Matteo Foundation, Pavia, Italy.
3 Unit of Microscopic and Ultrastructural Anatomy, Department of Medicine, University Campus Bio-Medico, Rome, Italy
4 Department of Biomedical and Neuromotorial Sciences, Alma Mater University, Bologna, Italy.
5 Department of Biomedical Engineering, Tufts University, Medford, MA, USA
Correspondence to: Prof.
Alessandra Balduini, MD, Department of Molecular Medicine, University
of Pavia, Pavia, Italy. Tel: +39 0382 502968. E-mail:
alessandra.balduini@unipv.it;
Prof. Annarita Migliaccio, PhD, Department of Biomedical and
Neuromotorial Sciences, Alma Mater University, Bologna, Italy. Tel: +39
051 2091547. E-mail:
annarita.migliaccio@unibo.it
Published: November 1, 2018
Received: August 15, 2018
Accepted: October 23, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018068 DOI
10.4084/MJHID.2018.068
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
In
Primary Myelofibrosis (PMF), megakaryocyte dysplasia/hyperplasia
determines the release of inflammatory cytokines that, in turn,
stimulate stromal cells and induce bone marrow fibrosis. The pathogenic
mechanism and the cells responsible for progression to bone marrow
fibrosis in PMF are not completely understood. This review article aims
to provide an overview of the crucial role of megakaryocytes in
myelofibrosis by discussing the role and the altered secretion of
megakaryocyte-derived soluble factors, enzymes and extracellular
matrices that are known to induce bone marrow fibrosis.
|
Introduction
Bone
marrow (BM) fibrosis is characterized by increased deposition of
reticulin fibres and in some cases collagen fibres.[1] There are a
number of hematologic and non-hematologic disorders that are associated
with increased BM fibrosis. In particular, reticulin fibres are
composed by type III collagen and may be evident in many benign
situations, including autoimmune and granulomatous diseases, and
different tumors, such as lymphoid neoplasms, myelodysplastic
syndromes, and acute myeloid leukemia. On the contrary, collagen
fibres, are composed of type I collagen and appear to be characteristic
of the advanced phases of myeloproliferative neoplasms (MPN), such as
primary myelofibrosis (PMF) or secondary myelofibrosis (MF) arising
from a pre-existing diagnosis of polycythemia vera (PV) or essential
thrombocythemia (ET).[1] The content of BM fibres in routine sections
of trephine biopsies of patients is usually demonstrated by
histochemical staining using silver impregnation for reticulin fibres
or by trichrome stains for collagen fibres.
Recent evidence has
shown that the amount of BM reticulin often exhibits no correlation to
disease severity, while the presence of collagen fibres is often
associated with more severe disease and a poorer prognosis.[2] The
exact pathogenesis of BM fibrosis is not fully understood. Aberrant
tyrosine kinase signaling is a common hallmark in MPNs and has been
shown to represent a key driver of the disease. The three currently
recognized driver mutations in PMF are JAK2 (Janus kinase 2; located on
chromosome 9p24), CALR (calreticulin; located on chromosome 19p13.2),
and MPL (myeloproliferative leukemia virus oncogene; located on
chromosome 1p34). The JAK2 V617F substitution of a valine for a
phenylalanine destabilizes the JH2 domain of JAK2 and causes loss of
the auto-inhibitory activity of this domain.[3] The most common MPL
mutations, W515L (tryptophan-to-leucine substitution) and W515K
(tryptophan-to-lysine substitution), cause both cytokine-independent
growth and hyper-TPO sensitivity.[4] JAK2V617F and MPL 515 mutations
are present in about 50% and 5% of PMF cases respectively, resulting in
permanent activation of the JAK/STAT signaling pathways and conferring in vitro
altered response of mutated clones to thrombopoietin (TPO) and other
cytokines.[3,5] In 2013 type 1 mutations (52-bp deletion) and type 2
mutations (5-bp insertion) were discovered in the Calreticulin gene.
These mutations determine a constitutive activation of the MPL receptor
through abnormal interaction with mutated calreticulin.[6,7]
Of
the myeloproliferative disorders, PMF has the worst overall prognosis
and morbidity.[8] Despite many significant advances in the treatment of
this disease, many aspects of its origin and progression remain poorly
understood. While recent understanding, on the pathogenic mechanisms in
hematopoietic stem cells (HSCs), provides an explanation for
myeloproliferation, several pieces of evidence clearly demonstrate that
other processes are involved in this disease than the simple
uncontrolled growth of mutant cells. In addition to BM fibrosis, the
malignant stem cells exit from the BM as the disease progresses, and
relocate in other hematopoietic organs, mostly the spleen and the
liver.[9] This leads to the enlargement of the spleen and liver that is
characteristic of this disease, causing significant morbidity.[10] In
PMF the pathogenesis of myelofibrosis appears to be intimately linked
with megakaryocyte (Mk) proliferation and differentiation.[11] Mks have
the primarily function to generate and release platelets in close
proximity of BM vasculature,[12] but they have also been shown to be
involved in the control of BM homeostasis through the generation of
signals that regulate HSC self-renewal and quiescence,[13,14] or
differentiation of others BM cell niche, such as plasma cells[15] or
osteoblasts.[16]
In PMF clusters of immature and necrotic Mks,
surrounded by fibrotic areas in the BM, suggests that improper or
premature release of their neat cargo of intracellular proteins unleash
the uncontrolled and disseminated fibrotic reaction driven by BM
stromal cells.[17] Noteworthy, Mks are very rich in cytokines, growth
factors, cross-linking enzymes and extracellular matrix proteins (ECM)
that are known to directly cause tissue fibrosis by stimulating stromal
cells to produce collagen or that physically participate to ECM
remodeling and BM scarring. Thus, here we will review what is known
about the potential contribution of Mks to the onset and progression of
BM fibrosis.
Megakaryocyte cargo in physiological thrombopoiesis
Mks
are unique, polyploid hematopoietic cells that are found only in
mammals, responsible for everyday production and release of millions of
platelets into the bloodstream.[18]
Megakaryopoiesis is mainly driven by TPO, although this cytokine may be dispensable for terminal megakaryocyte maturation in vitro.[19]
During the early stages of their differentiation, Mks become polyploid
through repeated DNA replication and endomitotic cycles without
cytokinesis.[20] At the end of maturation, the Mk cytoplasm becomes
very specialized with the development of a complex system of membranes,
called the demarcation membrane system (DMS), and three different types
of granules including lysosomes, dense granules and α-granules.[20] Proteins contained in the α-granules
(specialized secretory granules) can be synthesized or endocytosed.[21]
As in other cells, the cell-specific proteins are synthesized by
ribosomes on the rough endoplasmic reticulum and then packaged via the
Golgi apparatus into nascent granules.[22] Mk α-granules are the most abundant secretory organelles and contain a large variety of adhesive proteins, such as β-thromboglobulin,
CXCL4 (platelet factor 4, PF4), thrombospondin, fibronectin (FN), von
Willebrand factor (vWF) and P-selectin. In addition, anti-angiogenic
factors, endostatin and angiostatin, and pro-angiogenic factors such as
VEGF and SDF-1α, are involved in the regulation of lymphatic system development and vascular integrity promoted by α-granule.[23,24] Mk α-granules
contain additional growth factors for vascular repair, such as Platelet
Derived Growth Factor (PDGF), Transforming Growth Factor-β1
(TGFβ1), Epidermal Growth Factor (EGF) and Insulin Growth Factor
(IGF).[25] Furthermore, members of the metalloproteinases (MMP) and
tissue inhibitors of metalloproteinases (TIMP) family, which are
important factors in angiogenesis and tissue remodeling, are also
stored in Mk cytoplasm.[26] In addition, these granules have been shown
to contain several plasma proteins, such as fibrinogen,[27] Factor V,
albumin and immunoglobulin,[28] which are not synthesized by the cell
and are, therefore, endocytosed. Lastly, the pro-coagulant factors II,
V, XI and XIII, high molecular weight kininogens, the anti-fibrinolytic
factors plasminogen activator inhibitor-1, α2-antiplasmin
and carboxypeptidase B2, anti-coagulant factors, for example,
antithrombin, protein S, C1-inhibitor, TFPI and protease nexin 2 and
pro-fibrinolytic proteins such as plasminogen and plasmin have been all
localized in α-granules.[25]
Differently from α-granules,
secretory lysosomes contain distinct acid hydrolases, such as beta-
hexosaminidase, heparanase, elastase, and cathepsin D and E.[29]
Finally, Mk dense granules store small bioactive molecules, while α-granules are predominantly protein-packed. The cargo comprises nucleotides (e.g., ADP, ATP), polyphosphates, Ca2+ and Mg2+ cations, but also neurotransmitters and hormones, such as serotonin and histamine.[30]
How do megakaryocytes target and deliver their protein cargo within bone marrow milieu?
The constitutive in vitro secretion of both α-
and dense granules-derived bioactive molecules by developing Mks has
been previously demonstrated.[31,32] These functional experiments
revealed that secretion of intracellular Mk products governs autocrine
mechanisms that sustain cell development and platelet release.[33-36]
On the contrary, the physiological in vivo relevance of these autocrine
loops has been less explored. However, recent data shed new light on
the in vivo involvement of Mks in maintaining BM homeostasis though the
controlled release of targeted stimuli.[37] To this regard, conditional
ablation of Mks in mice resulted in increased BM HSC frequency and
cycling, suggesting that Mks normally restrain HSC proliferation
through the production of CXCL4 and TGF-β.[13,14]
Conversely, Mk production of fibroblast growth factor-1 (FGF-1) is
thought to play a key role in supporting HSC and osteoblast expansion
recovery following myeloablative therapy.[16] Therefore, these studies
raise the fundamental question: how is Mk compound exocytosis regulated
during physiologically or pathologically BM functions? Differently from
Mks, platelet activation is at the heart of the control of vascular
integrity.[38] During circulation, platelets are reactive to various
stimuli and release the materials stored in their specific
granules.[39] The extrusion of storage granules' content to the
platelet's environment occurs according to regulated secretion events:
movements of granules, apposition and fusion of granules and plasma
membranes.[40] This 'release reaction' is a key step of primary
hemostasis, but it participates also in inflammation, atherosclerosis,
antimicrobial host defense, wound healing, angiogenesis, and
malignancy.[41] Our current understanding of Mk/platelet secretion at
the molecular level is still insufficient to explain how the careful
balance between all the bioactive molecules released from granules,
under certain activation conditions, is achieved. One of the hypotheses
is that Mk/platelet granules are not uniform and may be differentially
packaged and thus released in a segregated manner following specific
stimuli.[42]
To this regard, Ma et al. observed that platelet
stimulation with specific protease-activated receptor- 1 (PAR-1) or
PAR-4 agonist resulted in the preferential release of VEGF or
endostatin (anti- and pro-angiogenic factors, respectively).[24,43]
More recently, a super-resolution immunofluorescence co-localization
analysis of 15 platelet α-granule cargoes failed to confirm any
functional co- clustering of these proteins.[44] Moreover, Zingariello
et al. demonstrated by immunoelectron microscopy that P-selectin and
von Willebrand factor (vWF) are co-localized within the same
intracellular α-granules in immature Mks. The two proteins, however,
are not co-localized in α- granules of mature Mks after wild type mice
treatment with TPO.[45] These results suggested that P- selectin and
vWF are associated in the Mk cytoplasm at early stages of maturation
but that they are routed into separate anti-angiogenic or
pro-angiogenic α-granule subtypes as these cells mature. Thus, the
differential association of vWF and P-selectin with anti-angiogenic or
pro-angiogenic factors suggest that a mechanism regulating the
sequential release of different α-granule subtypes may be involved in
Mk/platelet function during tissue repair.[45] Interestingly, Mks
derived from the GATA-1low mice,
which harbors a hypomorphic mutation that blocks Mk maturation and
displays BM fibrosis, showed reduced levels of expression of vWF and
displaced P-selectin on the demarcation membrane system.
Further,
the loss of alpha granules within BM Mks in a mouse model of Gray
Platelet Syndrome (GPS) induced a myelofibrotic phenotype.[46] In PMF
patients, ultrastructural abnormalities and variety in Mk-granules were
reported by Thiele et al. more than 25 years ago.[47] Platelets derived
from MPN patients showed several qualitative abnormalities, including
decreased alpha granules and mitochondria and also alterations of the
dense and tubular canalicular system.[48] Reduced levels of ADT, ATP
and serotonin content in dense granules and lower content of
beta-thromboglobulin (BTG) and platelet factor 4 (PF4) in alpha
granules were also described.[49,50] Thus, aberrant assembly and
secretion of Mk granules represent a potential mechanism of BM fibrosis
progression (Figure 1).
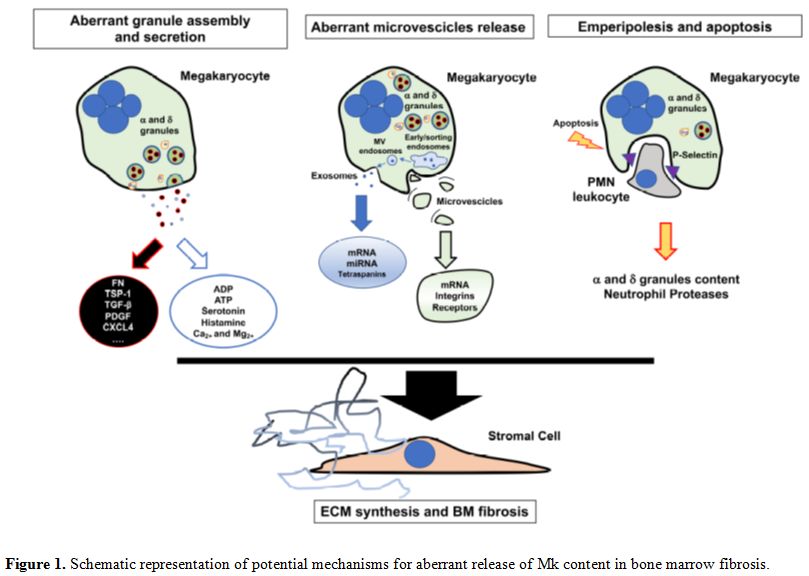 |
Figure 1. Schematic representation of potential mechanisms for aberrant release of Mk content in bone marrow fibrosis. |
A
second intriguingly hypothesis is that intracellular Mk/platelet
products may be delivered in the surrounding space by extracellular
vesicles (EVs). EVs are membrane-enclosed structures of varying size
(50-10,000 nm) released from cells to mediate both local and distant
intercellular communication.[51] EVs of various shapes and sizes have
been demonstrated in several body fluids, with substantial variation in
their structure, content and function.[52] There are three main types:
exosomes (50-100 nm), microparticles (200 nm-1 µm)
and apoptotic bodies. Protein, lipid and RNA components contribute to
cell-cell crosstalk at a short distance, in a paracrine or endocrine
manner via the bloodstream. In addition, they may transfer surface
receptors from one cell to another and deliver proteins, mRNA,
bioactive lipids, and even whole organelles (e.g., mitochondria) into
target cells.[53] It has been reported that two mechanisms used by
target cells to integrate EVs are cell endocytosis and membrane
fusion.[53] Recent studies of EVs in the BM have shown that these
vesicles serve to regulate hematopoiesis, participate in immune cell
activation, and hemostasis.[54,55] Several lines of evidence suggest
that EVs are involved in regulating BM function during homeostasis and
in response to injury, but also that hematological malignancies such as
leukemia, multiple myeloma or viral infections can exploit EVs
trafficking to reinforce tumor growth, chemotherapeutic resistance,
invasion and metastasis.[56,57] Platelet-derived vesicles were first
identified by electron microscopy over 50 years ago, but the definition
of their features and activities have only become a major focus of
interest in recent years.[58] Platelet-derived microparticles (PMPs)
are released from the platelet surface and are distinguished from
platelet exosomes, which are derived from endocytosis and released from
multivesicular endosomes.[59] PMPs may directly stimulate other cells
(e.g., hematopoietic cells, lymphocytes and endothelium),[60] transfer
platelet expressed receptors (e.g., CD41 or CXCR4) to the surface of
other cells,[61] and, in some situations, transfer mRNA, proteins, and
even infectious particles to the target cells. Interestingly, in
healthy donors, the majority of circulating CD41+ PMPs do not express
surface activation marker CD62P, suggesting that they do not originate
from activated platelets.[62] In a very elegant study, Flaumenhaft et
al. report that a significant number of circulating CD41+ MPs in
healthy individuals are derived directly from Mks.[63] Authors first
demonstrated via electron microscopy of spontaneous formation of
Mk-derived MPs (MkMPs) from cultured murine Mk and that these MkMPs
were different from PMPs. However, a functional role for MkMPs was not
revealed until a recent study, which documented a novel biological role
of MkMPs that are able to induce HSC differentiation towards the Mk
lineage without exogenous TPO stimulation.[64] In this paper, Jiang et
al. demonstrated that MkMPs, which are distinct from Mk exosomes,
target HSC with high specificity since they have no effect on other
BM-resident cells, such as mesenchymal stem cells, endothelial cells or
granulocytes. They showed that both endocytosis and membrane fusion
were responsible for the delivery of MkMP cargo to HSCs, and that MkMPs
attached to and entered HSCs preferentially through their uropods, with
CD54, CD11b, CD18 and CD43 being involved in target-cell
recognition.[64] Aside from the role of EVs in the Mk-HSC crosstalk in
the BM under physiological conditions, we can speculate that EV
trafficking may also play a distinct role in deregulated hematopoiesis
during fibrotic progression (Figure 1).
Interestingly, increased MP generation under high shear stress has been
reported in platelets in the presence of TPO,[65] a main trigger of BM
fibrosis in human and mice.
Megakaryocyte-derived pro-fibrotic cytokines in bone marrow fibrosis
In MPNs different mutations lead to myelofibrosis. The most frequent driver mutation in MPNs, JAK2
V617F, is found in 50-60% of PMF as well as 50-60% of ET, but in almost
all cases of PV.[66] It is now clear that the clinical phenotype of
myelofibrosis is a consequence both of primary clonal
myeloproliferation and secondary inflammation, characterized by
profound changes to BM stromal compartment and an atypical cytokine
storm.[67] Several evidences argue for an impaired microenvironment in
association with inflammation rather than one single genetic trigger:
1) MPNs (PV, ET, and PMF) are all characterized by a significant change
in the cytokine production mirrored by increased plasma levels of
several inflammatory cytokines (e.g., IL1, IL2, IL6, IL8, IL12, TNFα, and IFNγ),
growth factors (e.g., GM-CSF, G-CSF, HGF, PDGF, and EGF), and
angiogenic factors (e.g., VEGF);[68] 2) clinical evidences that chronic
inflammation is responsible of the constitutional symptoms which
negatively affect the quality of life of MPN patients;[69] 3) clinical
use of JAK inhibitors has confirmed that functional symptoms and
splenomegaly in patients were concomitant with a significant
increase in the plasma levels of many cytokines.[70]
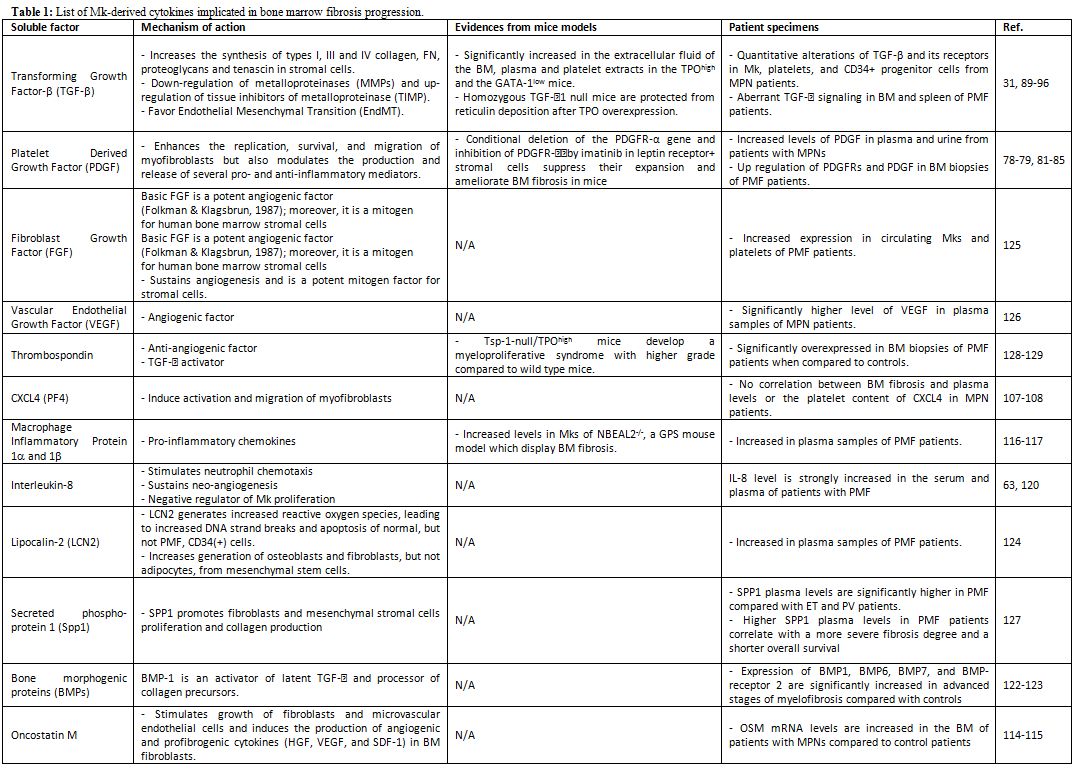 |
Table 1. List of Mk-derived cytokines implicated in bone marrow fibrosis progression. |
Platelet-Derived Growth Factor (PDGF).
PDGF is one of the first growth factors that has been implicated in the
role of Mk in development of BM fibrosis.[80] PDGF is produced by Mks
in the BM and is physiologically carried to the circulation in the
α-granules of the platelets to act at the site of tissue injury as a
mediator of tissue repair.[81] PDGF receptors (PDGFRs) are members of
the membrane tyrosin-kinase family, composed of the two subunits PDGFRα
and PDGFRβ, which form homo- or heterodimers. In the context of tissue
repair, the PDGF/PDGFR axis not only enhances the replication,
survival, and migration of myofibroblasts but also modulates the
production and release of several pro- and anti-inflammatory mediators
in fibrotic diseases.[82] Interestingly, PDGF has been reported to
increase the expression of the collagen cross-linking enzyme, Lysil
Oxidase (LOX), which in turn, oxidizes the PDGF receptor on smooth
muscle cells, fibroblasts, and Mks, enhancing the proliferation
signaling from this cytokine.[83,84] Ultimately, this loop has the
potential to further boost the fibrotic phenotype.[85]
Increased
levels of PDGF in plasma and urine from patients with MPNs have been
reported.[86,87] Further, Mks and erythroid precursors contained
increased levels of immunohistochemically detectable PDGF in BM
biopsies of PMF patients.[88] The expression of members of the PDGF
system in BM cells derived from PMF patients has been also investigated
by real-time RT-PCR.
Increased expression of PDGFs could be
demonstrated to be a feature of advanced fibrosis in PMF that is not
demonstrable in the pre-fibrotic phase of the disease.[88]
Differently
from their ligands, up-regulation of both PDGFRs during fibrotic
progression is more controversial. In normal BM PDGFRα appeared in
endothelial and endosteal cells in addition to strong labeling in Mks
and platelets. In contrast, PDGFRβ subunit marked perisinusoidal
stromal cells and adventitial fibrocytes of the larger vessels.
However, in PMF patients, Bedekovics et al. found that PDGFRβ
expression closely correlates with the grade of MF, while this was not
evident for PDGFRα.[90] On the contrary, Bock et al. reported a strong
up-regulation of the PDGFRα in patients with advanced myelofibrosis.[89]
Recently,
the involvement of the PDGF/PDGFR axis in BM fibrosis has been
definitively proven. Conditional deletion of the PDGFR-α gene and
inhibition of PDGFRα by imatinib in leptin receptor+ stromal cells was
shown to suppress their expansion and to ameliorate BM fibrosis in
mice.[91]
Transforming Growth Factor-β (TGF-β).
Among the abnormally expressed cytokines in PMF, TGF-β1 has received
attention due to its critical role in inducing fibrosis not only in BM,
but also in other organs.[92] TGF-β occurs in 3 isoforms: TGFβ1, TGFβ2
and TGFβ3. TGFβ1 is the most abundant of all these isoforms and
platelets, Mks and monocytes cells are sources of TGF-β production.[93]
TGFβ1 is secreted as latent protein and is stored in the extracellular
matrix. Reactive oxygen species, proteases, integrins and
thrombospondin-1 (TSP-1), convert the inactive latent complexes to the
active forms.
Once activated, TGFβ-1 induces BM fibrosis on one
hand, by increasing the synthesis of types I, III and IV collagen, FN,
proteoglycans and tenascin;[94] while on the other hand, by decreasing
matrix degradation through down-regulation of metalloproteinases
(MMPs), particularly MMP3, and up- regulation of tissue inhibitors of
metalloproteinase (TIMP), particularly TIMP-1.[95] However, effects of
TGF-β1
are not restricted to the stromal compartment and TGF-β1-mediated
changes to the BM niche remain to be fully elucidated. It is well known
that TGF-β has direct effects on hematopoietic cells by negatively
regulating granulocyte, erythroid, Mk and macrophage progenitor
proliferation.[96] Further, Erba et al. showed that release and
activation of TGF-β1 by Mks and platelets, forced endothelial cells
from the BM microvasculature of PMF patients, and mouse model of PMF,
to acquire a mesenchymal phenotype through Endothelial Mesenchymal
Transition (EndMT), during the development of fibrosis.[97]
Not
surprisingly, several groups reported on quantitative alterations of
TGF-β and its receptors in Mk, platelets, and CD34+ progenitor cells
from MPN patients and concluded that TGF-β was involved in
myelofibrosis and myeloproliferation.[31,98-100] In addition to
quantitative alterations, Ciaffoni et al., recently demonstrated
abnormalities in TGF-β1 signaling genes in the marrow and spleen of PMF
patients.[101] These alterations included genes of TGF-β1 signaling,
cell cycling, Hedgehog and p53 signaling and suggested a non-canonical
TGF-β1 signaling in marrow identifying, for the first time,
autoimmunity as a possible cause of BM fibrosis in PMF.[101] Among the
genes that predict the activation of the non-canonical TGF-β signaling,
expression level of the Jun gene was increased in BM of PMF patients.
Interestingly, over-expression of Jun was sufficient to induce
myelofibrosis, severe fibrosis in multiple organs and steatohepatosis
in mice.[102]
Moreover, the involvement of TGF-β in in vivo
mouse model has been deeply investigated. TGF-β was significantly
increased in the extracellular fluid of the BM, plasma and platelet
extracts in two widely MF studied mouse models, which include the TPOhigh and the GATA-1low mice.[103,104] To test directly the impact of TGF-β1 in the pathogenesis of MF, BM stem cells from homozygous TGF-β1 null (TGF-β1(-/-))
and wild-type littermates were infected with a retrovirus encoding the
murine TPO protein and engrafted into lethally irradiated wild type
hosts for long-term reconstitution. Differently from wild type mice,
none of the mice repopulated with TGF-β1(-/-)
cells showed deposition of reticulin fibres at any time during the
follow-up.[105] Consistently with patient data, alterations of TGF-β1,
Hedgehog, and p53 signaling pathways were identified in the BM of GATA-
1low mice model.[106] Inhibition of TGF-β1 signaling in these mice by
an inhibitor of the tyrosine kinase activity of TGF-β1 receptor type I,
led to restoration of normal Mk development, reduced fibrosis,
neoangiogenesis, and osteogenesis in the BM.[106] Based on these
consistent observations, TGF-β inhibition has become a potential
therapeutic strategy to decrease BM fibrosis in MPNs and is also being
investigated in several clinical and experimental scenarios.[107]
Cxcl-4 (Pf-4).
Cxcl-4, (C-X-C motif) ligand 4 (CXCL4) (also known as platelet factor 4
[PF4]), is one of the most abundant protein in the α-granules of
Mk/platelets (estimated micromolar concentration), together with
CXCL7.[108] This 70 a.a., cationic, lysine-rich, 7.8-kDa chemokine, is
mainly synthesized by Mks, and comprises 2%-3% of the releasate from
agonist-activated platelets. Once secreted, CXCL4 avidly binds to
glycosaminoglycans, but only a splice-variant of the human chemokine
receptor CXCR3 (CXCR3B), which is not present in mice, and LDLR90 have
been identified as high-affinity receptors.[109] In contrast, the
specific receptor for CXCL4 has not yet been identified in mice.
However, in some circumstances, CXCL4 can interact with other
chemokines (e.g. CCL5) and thereby modulate their effects on target
cells. A central role of platelet-derived CXCL4 was demonstrated in
solid organs. In vivo, mice
lacking CXCL4 are significantly protected from severe liver fibrosis,
demonstrating the pro-fibrotic phenotype of this chemokine and that its
effects in mice are indeed mediated by other receptors than CXCR3.[110]
In addition, it was shown that CXCL4 is secreted not only by activated
platelets, but also by plasmacytoid dendritic cells and fibroblasts in
systemic sclerosis.[111] Thus, these studies further involve
Mks/platelets to pro- inflammatory and pro-fibrotic programmes in
fibrosis. Interestingly, more than 30 years ago, Burstein et al. linked
CXCL4 to myelofibrosis, by suggesting that abnormal Mks stimulate the
proliferation of fibrosis-driving fibroblasts though the release of
CXCL4.[112] However, no correlation was seen between BM fibrosis and
plasma levels or the platelet content of CXCL4 in the same study.[112]
Recently, Schneider et al. using a mouse model with genetic fate
tracing in vivo, provided
evidence that Gli1+ cells are key players in the initiation and
progression of BM fibrosis and that Mk-derived CXCL4 was necessary and
sufficient to induce the migration of Gli1+ stromal cells and their
myofibroblastic differentiation.[113] In these experiments, CXCL4 was
shown to induced myofibroblast differentiation of Gli1+ cells
comparable to induction with TGF-β, a known stimulus for
differentiation of MSCs into myofibroblasts.[113]
Other cytokines.
Oncostatin M (OSM), is a pleiotropic cytokine belonging to the
interleukin-6 (IL-6) family.[114] Produced mainly by activated T cells
and monocytes, OSM can elicit different biological effects, depending
on the cell type. OSM acts through two types of receptors. The type I
OSM receptor is composed of gp130 and the leukemia inhibitory factor
(LIF) receptor β-subunit (LIFR), and the type II OSM receptor is
composed of gp130 and the OSM-specific receptor β- subunit (OSMR).[115]
OSM has emerged as an important cytokine in the control of
hematopoiesis. Transplantation experiments with OSM-deficient mice have
shown that OSM stimulates stromal cells as well as hematopoietic
progenitors and is required for the proper generation and maintenance
of microenvironment in the BM.[116,117] Noteworthy, BM Mks express
substantial amounts of OSM,[118] and OSM has been reported to behave as
a megakaryocytic maturation factor in vitro and to augment platelet production in vivo.[119]
Within the context of myelofibrosis, JAK2 V617F mutation promotes
expression of OSM in neoplastic myeloid cells and, consequently, OSM
mRNA levels are increased in the BM of patients with MPNs compared to
control patients.[120] Mechanistically, OSM secreted by JAK2V617F+
cells stimulated growth of fibroblasts and endothelial cells by
sustaining the production of angiogenic and pro-fibrogenic
cytokines.[120]
Aberrant packaging of α-granule-specific
proteins is supposed to trigger myelofibrosis in patients with GPS.
Using a Nbeal2-/- murine model of GPS, Guerrero et al. demonstrated
that BM Mks from these mice were enriched in a restricted set of
chemokines transcripts, namely CCL3 and CCL4, which encode macrophage
inflammatory protein (MIP) 1α and 1β, respectively, well-known pro- inflammatory chemokines increased in PMF.[46] A peculiar role for MIP 1α in sustaining osteoblasts proliferation in MPN mice model has been also proposed.[121]
Interleukin
8 (IL-8) is a member of the family of chemokines related by a CXC
motif. It binds to CXC chemokine receptor 1 (CXCR1) and 2 (CXCR2).[122]
It is produced by several cell types, including Mks[123] and exhibits
many biological functions in inflammation, HSC proliferation and
mobilization and neo-angiogenesis. Increased levels of IL-8 were found
in serum[124] and plasma[68] of patients with PMF. Additionally, IL-8
and its receptors were reported to be involved in PMF-altered Mk
growth.[124] Finally, rodents lack a direct homologue of IL-8, but the
chemokines CXCL1/KC, CXCL2/MIP-2, and CXCL5-6/LIX are regarded as
functional homologues of IL-8.[125]
Finally, Mks were
repeatedly identified as the main cellular source of an
increasing list of cytokines, which show higher plasma levels in PMF
patients, and that are individually involved in the promotion of
myelofibrosis. This list further includes bone morphogenic proteins
(e.g. BMP- 1[126], BMP-2, -4, and -6[127]), Lipocalin-2 (LCN-2),[128]
Fibroblasts Growth Factor (FGF),[129] Vascular Endothelial Growth
Factor (VEGF),[130] Secreted Phospho Protein-1 (SPP1)[131] and
Thrombospondin- 1 (TSP-1).[132,133]
Megakaryocyte expression of extracellular matrices and cross-linking enzymes in bone marrow fibrosis
Deregulated
extracellular matrix (ECM) dynamics in terms of amount, composition and
topography is a hallmark of BM fibrosis.[1] This in turn potentiates
the oncogenic effects of growth factor signaling pathways and alters
cell behaviors during fibrosis progression. ECM components are not
solely expressed by stromal cells, several evidences suggest that Mks
may directly influence the biochemical properties and architecture of
BM ECM both in physiological and pathological conditions.[134] It is
known that Mks can secrete various ECM components which are supposed to
sustain Mk maturation and platelet release by creating a regulatory
niche within the BM environment.[135] Mks express different collagen
types (e.g., III, IV), glycoproteins (e.g., Fibronectin and
Thrombospondin) and proteoglycans. Interestingly, TPO has been recently
recognized as a pivotal regulator of this new Mk function, by inducing
TGF-β1 release and consequent activation of TGF-β downstream signaling pathways, both in vitro and in vivo.[136]
This activation led to a dose dependent increase of ECM component
synthesis by Mks, which was reverted upon incubation with JAK and
TGF-β1 receptor specific inhibitors.[136]In
parallel with ECM secretion, Mks express several modifiers of ECM
structure. Factor XIII-A is synthesized by Mks and both protein and
mRNA are packaged into the cytoplasm of forming platelets.[137] Factor
XIII-A belongs to transglutaminases, a class of calcium ion-dependent
enzymes that catalyze an acyl transfer reaction in which y-carboxamide
groups of peptide-bound glutaminyl residues are acyl donors and primary
amine including the δ-amino
group of peptide-bound lysyl residues, are acyl acceptors. By this
reaction, transglutaminases catalyze the formation of δ-(y-
glutamy1) lysine linkages between proteins. Thus, based on these
properties, the potential role of FXIII-A in the BM environment may
consist in the cross-link of extracellular fibrillar FN matrix with
collagen.[138,139]LOX
is a copper-dependent amine oxidase that catalyzes oxidative
deamination of lysine and hydroxylysine residues on collagen and
elastin, leading to cross-linking within these proteins and changes in
ECM elasticity. Eliades et al., detected LOX expression in
diploid-tetraploid Mks, but scarce traces in polyploid Mks and
identified a peculiar role for this enzyme in BM fibrosis.[84] They
found that in the GATA-1low mouse
model, which is characterized by increased frequency of low ploidy Mks
and extensive matrix of fibres, LOX was abundantly expressed by low
ploidy Mks. More importantly, administration of β-aminopropionitrile (a
LOX inhibitor) to the GATA-1low mice
inhibited the progression of myelofibrosis. Consistently, human
platelets and Mks from patients with PMF overexpress LOX and show
higher adhesion to collagen that is dependent on LOX activity.[140]In
addition to cross-linking enzymes, also ECM degradation directly
impacts cell behavior and migration. Metalloproteinases (MMPs) are a
family of zinc-dependent endopeptidases and function in remodeling the
ECM by its ability to degrade and cleave ECM components with wide
substrate specificities.[141] Once activated, the MMPs are subject to
inhibition by the tissue inhibitors of metalloproteinases (TIMPs) that
bind MMPs non-covalently and counteract their proteolytic activity.Mks
synthetize several MMPs, particularly gelatinases MMP-2 and MMP-9.[142]
Moreover, transcripts for MMP-1, 11, 14, 15, 17, 19, 24 and 25 have
also been identified.[26] Conversely, biosynthesis of TIMPs 1-4 in
Mks/platelets intervenes in excessive tissue remodeling.[143] It is
suggested that BM fibrosis in PMF results from enhanced TIMP and
decreased MMP activities. In particular, TIMP-1 (both the total,
complex and the free form) is significantly increased in MPNs, while
MMP-3 is significantly decreased, and levels of MMP-2 and MMP-9 are not
different from control values.[95,144] Further, membrane type 1-MMP
(MMP-14) was found overexpressed by up to 80-fold in advanced stages of
fibrosis, and Mks and endothelial cells were unmasked as the major
cellular source.[145] By contrast, a significantly higher expression of
neutrophil collagenase (MMP-8) was encountered in the pre-fibrotic
stages of PMF. Although the JAK-STAT signaling pathway is directly
involved in the regulation of genes encoding MMPs, the altered
expression of MMPs seem not influenced by the JAK2 mutation status but predominantly related to the stage of disease.[145] Direct Megakaryocyte-cell interactions in the context of bone marrow fibrosis
In
addition to secretory events, one more pathophysiological mechanism
operating in the development of myelofibrosis is the abnormal
interaction of Mks with cell components of the BM (Figure 1).
Selectins (CD62L, CD62P) and Mk glycoproteins (CD41a, CD42b) were
demonstrated to mediate Mk-fibroblast interactions in human BM and to
increase fibroblast growth.[146]Abnormalities
in mesenchymal stem cells derived from PMF patients were reported to
alter the ability of these cells to support Mk differentiation in vitro.[147-149]
Further, a pathological interaction, between polymorphonuclear (PMN)
leukocytes and Mk, correlated with MF development, has been also
proposed.[17] Emperipolesis is the random passage of the different
types of BM cells through Mk intracellular space. The phenomenon is
strongly increased in BM of patients with MPN disorders.[150] Schmitt
et al., first showed both in the BM of patients with PMF, and in the TPOhigh
murine model, abnormal subcellular P-selectin distribution, which
appeared to correlate with excessive and pathological emperipolesis of
PMN leukocytes within Mk.[150] This abnormal interaction was considered
the main cause of the destruction of Mk storage organelles and leakage
of α-granular
contents into the BM microenvironment.[151] As in patients, a similar
pathologic neutrophil emperipolesis was detected in the GATA-1low
mouse model of myelofibrosis.[152] In BM Mk of these mice, P-selectin,
although normally expressed, was found frequently associated with the
demarcation membrane system (DMS) instead of within granules. In
addition, pathologic Mks were surrounded by myeloperoxidase-positive
neutrophils, some of which appeared in the process to establish contact
with Mks by fusing their membrane with those of the DMS. Quantification
of this process revealed that 34% (in BM) of GATA-1(low)
Mks contained 1 to 3 neutrophils embedded in a vacuolated cytoplasm.
The neutrophil-embedded GATA-1(low) Mks displayed morphologic features
compatible with those of cells dying from para-apoptosis, confirming
the hypothesis that emperipolesis sustains myelofibrosis by driving the
release of fibrogenic Mk cytokines and neutrophil proteases in the BM
microenvironment.[152] Moreover, abnormal localization of P-selectin in
Mks and platelets, induced by the GATA-1(low)
mutation, was further involved in the pathological interactions of
circulating platelets with leucocytes, responsible for the increased
presence of thrombosis seen in these mice,[153] as well as, in the
promotion of extramedullary hematopoiesis.[154] Consistently, high rate
of emperipolesis is detectable in BM biopsies of patients with GPS, a
rare inherited bleeding disorder characterized by deficiency of
platelet α-granules, macrothrombocytopenia and marrow fibrosis.[155] Is the pro-fibrotic role of Megakaryocytes/ platelets restricted to the bone marrow?
New
discoveries in the field of thrombopoiesis and platelet roles have
revealed unprecedented features of the Mk/Platelet lineage that open
new avenues in the study of these cells, particularly in diseased
conditions. Bioactive mediators, stored in platelets, have been
implicated in fibrotic conditions that target solid organs, rather than
BM (Table 2).
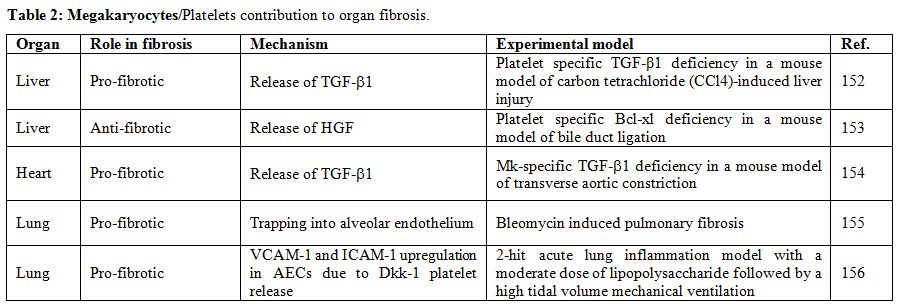 |
Table 2. Megakaryocytes/Platelets contribution to organ fibrosis. |
A
large amount of experimental evidence implies that platelets
participate in the liver fibrotic process mainly by releasing pro-
fibrotic mediators. Using mice carrying a Mk/platelet-specific targeted
conditional deletion of the TGF-β1 gene (PF4CreTgfβ1f/f),
Ghafoory et al. demonstrated that platelet TGF-β1 deficiency decreases
liver fibrosis in a mouse model of carbon tetrachloride (CCl4)-induced
liver injury.[156] However, there is also evidence that platelets under
certain circumstances may have a protective role against liver
fibrosis. To this regard, thrombocytopenic mice, with selective
disruption of the anti-apoptotic gene Bcl-xL, were shown to be more
prone to liver fibrosis by bile duct ligation compared to their wild
type counterparts.[157] The authors, suggested that the anti-fibrotic
Hepatocyte Growth Factor (HGF) released from activated platelets in
liver, attenuated the expression of collagen in hepatic stellate cells,
the key cell type in liver fibrosis.[157] Additionally, Mk-specific
disruption of the TGF-β1
gene resulted in mice protection from cardiac hypertrophy, fibrosis,
and systolic dysfunction in response to transverse aortic constriction,
suggesting that platelet profibrotic behavior is not solely restricted
to the liver.[158] Similarly, evidence has been accumulated implicating
platelets in the pathogenesis of interstitial lung fibrosis in several
animal models. Piguet et al. found that trapping of platelets in
contact with the alveolar endothelium of the lungs after bleomycin
injection was increased and correlated with the deposition of
collagen.[159] The authors suggested that this could represent not only
a simple correlation but also a potential pathological mechanism that
links platelets and pulmonary fibrosis. Interestingly, in a recent
study platelets were shown to promote acute lung injury through the
massive release of the Wnt/β-catenin inhibitor Dickkopf-1 (Dkk-1) from
their α-granules, leading to increased expression of vascular cell
adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1
(ICAM-1) on the surface of alveolar epithelial cells (AECs) and
abnormal macrophage/neutrophils interaction with AECs.[160] In addition
to the direct involvement of platelets in fibrosis of solid organs, the
potential contribution of Mks to organ fibrosis still needs to be
uncovered. It is becoming increasingly clear that Mks transfer unique
genetic codes to platelets, and that environmental changes can alter
transcriptional, translational, and post-translational processes in
Mks, affecting the genetic code of platelets in circulation.[161]
Change in Mk-platelet transcriptional axis is a dynamic process,
especially in disease situations, and can rapidly affect the platelet
repertoire of messenger RNAs (mRNAs), microRNAs (miRNAs), and proteins
that contribute to their primary and alternative functions. Freishtat
et al., revealed for the first time, that a de novo transcriptome is
imparted to platelets by BM residing Mks during sepsis.[162] Septic Mks
produce platelets with acutely altered mRNA profiles, and these
platelets mediate lymphotoxicity via the potent cytotoxic serine
protease, granzyme B.[162] Similarly, in the context of cancer, Mks of
tumor-bearing mice endocytose circulating thrombospondin-1 (TSP-1) and
increase its synthesis to produce platelets with elevated levels of
TSP-1, one of the most potent angiogenesis inhibitors. These
TSP-1-enriched platelets were shown to adhere to tumors and to act as
potent inhibitors of angiogenesis and cancer growth.[163] Thus, similar
changes may occur in fibrotic conditions, but this has not been
demonstrated yet. Conclusions
In
this review, we summarized the involvement of the Mk lineage in the
development of BM fibrosis. We now know that, in addition to genetic
triggers, BM fibrosis is sustained by the intramedullary release of
cytokines that are responsible for the abnormal activation of stromal
cells, resulting in extensive deposits of reticulin and collagens. Mks
are supposed to constitute the main source of these reactive cytokines.
Abnormal Mk differentiation, apoptosis and emperipolesis were all
proposed as major mechanisms for the enhanced release of cytokines with
a fibrogenic potential. Unfortunately, mechanisms underlying Mk
secretion, their relationships with other BM lineages and their
functional activities in physiological conditions as well as during
myelofibrosis progression, are not well understood to date. The first
attempt to directly target the Mk lineage was shown, recently, to
revert the disease in both Jak2V617F and MPLW515L mice models. Using a
small molecule, the AURKA inhibitor MLN8237, that induce Mk
polyploidization, differentiation, and subsequent apoptosis, the
Crispino’s group demonstrated that the pharmacological induction of Mk
maturation was beneficial in terms of reduced burden of immature Mks
and amelioration of PMF features, including BM fibrosis.[164] Thus,
developing drugs able to re-establish Mk normal function may represent
a new strategy to treat the disease and, at the same time, to
understand its pathogenic mechanisms (Figure 2).
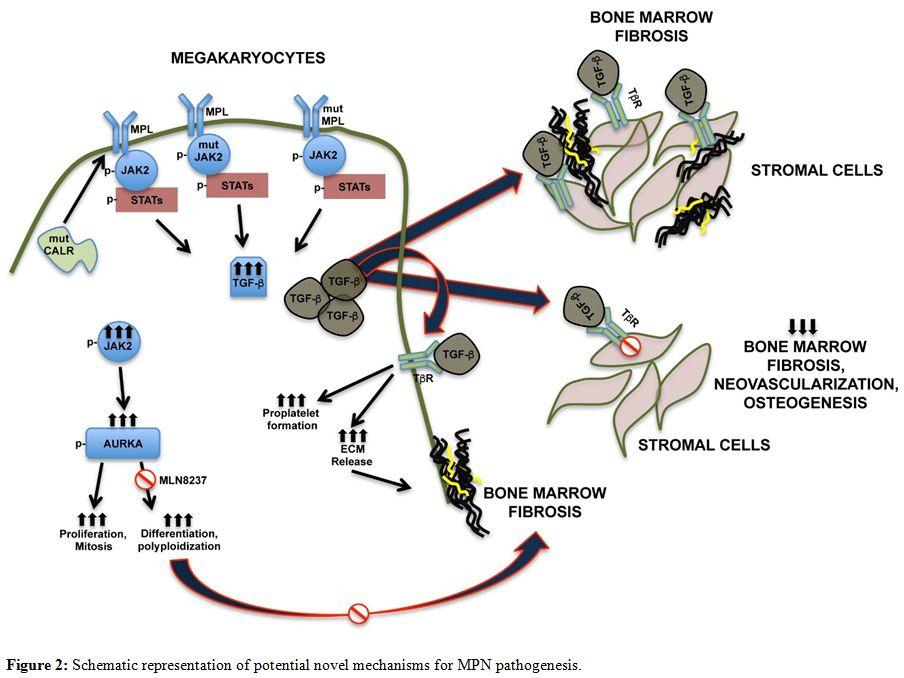 |
Figure 2. Schematic representation of potential novel mechanisms for MPN pathogenesis. |
Acknowledgments
This paper was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC IG 2016 18700, AIRC; Milano, Italy).
References
- Kuter DJ, Bain B, Mufti G, Bagg A, Hasserjian RP.
Bone marrow fibrosis: pathophysiology and clinical significance of
increased bone marrow stromal fibres. Br J Haematol.
2007;139(3):351-362. https://doi.org/10.1111/j.1365-2141.2007.06807.x
PMid:17910625
- Gianelli U, Fiori S, Cattaneo D, et al.
Prognostic significance of a comprehensive histological evaluation of
reticulin fibrosis, collagen deposition and osteosclerosis in primary
myelofibrosis patients. Histopathology. 2017;71(6):897-908.
https://doi.org/10.1111/his.13309 PMid:28710830
- James
C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading
to constitutive signalling causes polycythaemia vera. Nature.
2005;434(7037):1144-1148. https://doi.org/10.1038/nature03546
PMid:15793561
- Pikman Y, Lee BH, Mercher T, et al.
MPLW515L is a novel somatic activating mutation in myelofibrosis with
myeloid metaplasia. PLoS Med. 2006;3(7):e270.
https://doi.org/10.1371/journal.pmed.0030270 PMid:16834459
PMCid:PMC1502153
- Pardanani AD, Levine RL, Lasho T, et
al. MPL515 mutations in myeloproliferative and other myeloid disorders:
a study of 1182 patients. Blood. 2006;108(10):3472-3476.
https://doi.org/10.1182/blood-2006-04-018879 PMid:16868251
- Klampfl
T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of
calreticulin in myeloproliferative neoplasms. N Engl J Med.
2013;369(25):2379-2390. https://doi.org/10.1056/NEJMoa1311347
PMid:24325356
- Nangalia J, Massie CE, Baxter EJ, et al.
Somatic CALR mutations in myeloproliferative neoplasms with nonmutated
JAK2. N Engl J Med. 2013;369(25):2391-2405.
https://doi.org/10.1056/NEJMoa1312542 PMid:24325359 PMCid:PMC3966280
- Vannucchi
AM, Guglielmelli P, Tefferi A. Advances in understanding and management
of myeloproliferative neoplasms. CA Cancer J Clin. 2009;59(3):171-191.
https://doi.org/10.3322/caac.20009 PMid:19369682
- Mesa
RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post
polycythemia vera myelofibrosis (post-PV MF), post essential
thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP):
Consensus on terminology by the international working group for
myelofibrosis research and treatment (IWG-MRT). Leuk Res.
2007;31(6):737-740. https://doi.org/10.1016/j.leukres.2006.12.002
PMid:17210175
- Cervantes F, Dupriez B, Pereira A, et al.
New prognostic scoring system for primary myelofibrosis based on a
study of the International Working Group for Myelofibrosis Research and
Treatment. Blood. 2009;113(13):2895-2901.
https://doi.org/10.1182/blood-2008-07-170449 PMid:18988864
- Ciurea
SO, Merchant D, Mahmud N, et al. Pivotal contributions of
megakaryocytes to the biology of idiopathic myelofibrosis. Blood.
2007;110(3):986-993. https://doi.org/10.1182/blood-2006-12-064626
PMid:17473062 PMCid:PMC1924766
- Avecilla ST, Hattori K,
Heissig B, et al. Chemokine-mediated interaction of hematopoietic
progenitors with the bone marrow vascular niche is required for
thrombopoiesis. Nat Med. 2004;10(1):64-71.
https://doi.org/10.1038/nm973 PMid:14702636
- Bruns
I, Lucas D, Pinho S, et al. Megakaryocytes regulate hematopoietic stem
cell quiescence through CXCL4 secretion. Nat Med.
2014;20(11):1315-1320. https://doi.org/10.1038/nm.3707 PMid:25326802
PMCid:PMC4258871
- Zhao M, Perry JM, Marshall H, et al.
Megakaryocytes maintain homeostatic quiescence and promote post-injury
regeneration of hematopoietic stem cells. Nat Med.
2014;20(11):1321-1326. https://doi.org/10.1038/nm.3706
PMid:25326798
- Winter O, Moser K, Mohr E, et al.
Megakaryocytes constitute a functional component of a plasma cell niche
in the bone marrow. Blood. 2010;116(11):1867-1875.
https://doi.org/10.1182/blood-2009-12-259457 PMid:20538807
- Olson
TS, Caselli A, Otsuru S, et al. Megakaryocytes promote murine
osteoblastic HSC niche expansion and stem cell engraftment after
radioablative conditioning. Blood. 2013;121(26):5238-5249.
https://doi.org/10.1182/blood-2012-10-463414 PMid:23667055
PMCid:PMC3695366
- Schmitt A, Jouault H, Guichard J,
Wendling F, Drouin A, Cramer EM. Pathologic interaction between
megakaryocytes and polymorphonuclear leukocytes in myelofibrosis.
Blood. 2000;96(4):1342-1347. PMid:10942376
- Kaushansky
K. The molecular mechanisms that control thrombopoiesis. J Clin Invest.
2005;115(12):3339-3347. https://doi.org/10.1172/JCI26674 PMid:16322778
PMCid:PMC1297257
- Shivdasani RA, Rosenblatt MF,
Zucker-Franklin D, et al. Transcription factor NF-E2 is required for
platelet formation independent of the actions of thrombopoietin/MGDF in
megakaryocyte development. Cell. 1995;81(5):695-704.
https://doi.org/10.1016/0092-8674(95)90531-6
- Patel
SR, Hartwig JH, Italiano JE. The biogenesis of platelets from
megakaryocyte proplatelets. J Clin Invest. 2005;115(12):3348-3354.
https://doi.org/10.1172/JCI26891 PMid:16322779 PMCid:PMC1297261
- Blair
P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical
correlates. Blood Rev. 2009;23(4):177-189.
https://doi.org/10.1016/j.blre.2009.04.001 PMid:19450911
PMCid:PMC2720568
- Maynard DM, Heijnen HF, Gahl WA,
Gunay-Aygun M. The α-granule proteome: novel proteins in normal and
ghost granules in gray platelet syndrome. J Thromb Haemost.
2010;8(8):1786-1796. https://doi.org/10.1111/j.1538-7836.2010.03932.x
PMid:20524979 PMCid:PMC2953603
- Stellos K, Langer H,
Daub K, et al. Platelet-derived stromal cell-derived factor-1 regulates
adhesion and promotes differentiation of human CD34+ cells to
endothelial progenitor cells. Circulation. 2008;117(2):206-215.
https://doi.org/10.1161/CIRCULATIONAHA.107.714691 PMid:18086932
- Battinelli
EM, Markens BA, Italiano JE. Release of angiogenesis regulatory
proteins from platelet alpha granules: modulation of physiologic and
pathologic angiogenesis. Blood. 2011;118(5):1359-1369.
https://doi.org/10.1182/blood-2011-02-334524 PMid:21680800
PMCid:PMC3152500
- Coppinger JA, Cagney G, Toomey S, et
al. Characterization of the proteins released from activated platelets
leads to localization of novel platelet proteins in human
atherosclerotic lesions. Blood. 2004;103(6):2096-2104.
https://doi.org/10.1182/blood-2003-08-2804 PMid:14630798
- Cecchetti
L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes
differentially sort mRNAs for matrix metalloproteinases and their
inhibitors into platelets: a mechanism for regulating synthetic events.
Blood. 2011;118(7):1903-1911.
https://doi.org/10.1182/blood-2010-12-324517 PMid:21628401
PMCid:PMC3158719
- Handagama P, Scarborough RM, Shuman
MA, Bainton DF. Endocytosis of fibrinogen into megakaryocyte and
platelet alpha-granules is mediated by alpha IIb beta 3 (glycoprotein
IIb-IIIa). Blood. 1993;82(1):135-138. PMid:8391871
- George
JN. Platelet immunoglobulin G: its significance for the evaluation of
thrombocytopenia and for understanding the origin of alpha-granule
proteins. Blood. 1990;76(5):859-870. PMid:2203482
- Ciferri
S, Emiliani C, Guglielmini G, Orlacchio A, Nenci GG, Gresele P.
Platelets release their lysosomal content in vivo in humans upon
activation. Thromb Haemost. 2000;83(1):157-164.
https://doi.org/10.1055/s-0037-1613772 PMid:10669170
- McNicol
A, Israels SJ. Platelet dense granules: structure, function and
implications for haemostasis. Thromb Res. 1999;95(1):1-18.
https://doi.org/10.1016/S0049-3848(99)00015-8
- Badalucco
S, Di Buduo CA, Campanelli R, et al. Involvement of TGFβ1 in autocrine
regulation of proplatelet formation in healthy subjects and patients
with primary myelofibrosis. Haematologica. 2013;98(4):514-517.
https://doi.org/10.3324/haematol.2012.076752 PMid:23403314
PMCid:PMC3659980
- Balduini A, Di Buduo CA, Malara A, et
al. Constitutively released adenosine diphosphate regulates proplatelet
formation by human megakaryocytes. Haematologica. 2012;97(11):1657-
1665. https://doi.org/10.3324/haematol.2011.059212 PMid:22689668
PMCid:PMC3487437
- Saulle E, Guerriero R, Petronelli A,
et al. Autocrine role of angiopoietins during megakaryocytic
differentiation. PLoS One. 2012;7(7):e39796.
https://doi.org/10.1371/journal.pone.0039796 PMid:22792187
PMCid:PMC3391299
- Lambert MP, Rauova L, Bailey M,
Sola-Visner MC, Kowalska MA, Poncz M. Platelet factor 4 is a negative
autocrine in vivo regulator of megakaryopoiesis: clinical and
therapeutic implications. Blood. 2007;110(4):1153-1160.
https://doi.org/10.1182/blood-2007-01-067116 PMid:17495129
PMCid:PMC1976471
- Nagata Y, Yoshikawa J, Hashimoto A,
Yamamoto M, Payne AH, Todokoro K. Proplatelet formation of
megakaryocytes is triggered by autocrine-synthesized estradiol. Genes
Dev. 2003;17(23):2864-2869. https://doi.org/10.1101/gad.1128003
PMid:14665668 PMCid:PMC289146
- Casella I, Feccia T,
Chelucci C, et al. Autocrine-paracrine VEGF loops potentiate the
maturation of megakaryocytic precursors through Flt1 receptor. Blood.
2003;101(4):1316-1323. https://doi.org/10.1182/blood-2002-07-2184
PMid:12406876
- Malara A, Abbonante V, Di Buduo CA, Tozzi
L, Currao M, Balduini A. The secret life of a megakaryocyte: emerging
roles in bone marrow homeostasis control. Cell Mol Life Sci.
2015;72(8):1517-1536. https://doi.org/10.1007/s00018-014-1813-y
PMid:25572292 PMCid:PMC4369169
- Clemetson KJ. Platelets
and primary haemostasis. Thromb Res. 2012;129(3):220-224.
https://doi.org/10.1016/j.thromres.2011.11.036 PMid:22178577
- Eckly
A, Rinckel JY, Proamer F, et al. Respective contributions of single and
compound granule fusion to secretion by activated platelets. Blood.
2016;128(21):2538-2549. https://doi.org/10.1182/blood-2016-03-705681
PMid:27625359
- Rendu F, Brohard-Bohn B. The platelet
release reaction: granules' constituents, secretion and functions.
Platelets. 2001;12(5):261-273.
https://doi.org/10.1080/09537100120068170 PMid:11487378
- Golebiewska
EM, Poole AW. Platelet secretion: From haemostasis to wound healing and
beyond. Blood Rev. 2015;29(3):153-162.
https://doi.org/10.1016/j.blre.2014.10.003 PMid:25468720
PMCid:PMC4452143
- Italiano JE, Richardson JL, Patel-Hett
S, et al. Angiogenesis is regulated by a novel mechanism: pro- and
antiangiogenic proteins are organized into separate platelet alpha
granules and differentially released. Blood. 2008;111(3):1227-1233.
https://doi.org/10.1182/blood-2007-09-113837 PMid:17962514
PMCid:PMC2214735
- Ma L, Perini R, McKnight W, et al.
Proteinase-activated receptors 1 and 4 counter-regulate endostatin and
VEGF release from human platelets. Proc Natl Acad Sci U S A.
2005;102(1):216- 220. https://doi.org/10.1073/pnas.0406682102
PMid:15615851 PMCid:PMC544057
- Kamykowski J, Carlton P,
Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals
little functional coclustering of proteins within platelet α-granules.
Blood. 2011;118(5):1370-1373.
https://doi.org/10.1182/blood-2011-01-330910 PMid:21622648
- Zingariello
M, Fabucci ME, Bosco D, et al. Differential localization of P-selectin
and von Willebrand factor during megakaryocyte maturation. Biotech
Histochem. 2010;85(3):157-170.
https://doi.org/10.3109/10520290903149612 PMid:20426698
PMCid:PMC3700322
- Guerrero JA, Bennett C, van der Weyden
L, et al. Gray platelet syndrome: proinflammatory megakaryocytes and
α-granule loss cause myelofibrosis and confer metastasis resistance in
mice. Blood. 2014;124(24):3624-3635.
https://doi.org/10.1182/blood-2014-04-566760 PMid:25258341
- Thiele
J, Kuemmel T, Sander C, Fischer R. Ultrastructure of bone marrow tissue
in so-called primary (idiopathic) myelofibrosis-osteomyelosclerosis
(agnogenic myeloid metaplasia). I. Abnormalities of megakaryopoiesis
and thrombocytes. J Submicrosc Cytol Pathol. 1991;23(1):93- 107.
PMid:2036630
- Raman BK, Van Slyck EJ, Riddle J, Sawdyk
MA, Abraham JP, Saeed SM. Platelet function and structure in
myeloproliferative disease, myelodysplastic syndrome, and secondary
thrombocytosis. Am J Clin Pathol. 1989;91(6):647-655.
https://doi.org/10.1093/ajcp/91.6.647 PMid:2524965
- Sacchi
S, Curci G, Piccinini L, et al. Platelet alpha-granule release in
chronic myeloproliferative disorders with thrombocytosis. Scand J Clin
Lab Invest. 1986;46(2):163-166.
https://doi.org/10.3109/00365518609083653 PMid:2424075
- Holme S, Murphy S. Platelet abnormalities in myeloproliferative disorders. Clin Lab Med. 1990;10(4):873-888. PMid:2272179
- Muralidharan-Chari
V, Clancy JW, Sedgwick A, D'Souza-Schorey C. Microvesicles: mediators
of extracellular communication during cancer progression. J Cell Sci.
2010;123(Pt 10):1603-1611. https://doi.org/10.1242/jcs.064386
PMid:20445011 PMCid:PMC2864708
- Raposo G, Stoorvogel W.
Extracellular vesicles: exosomes, microvesicles, and friends. J Cell
Biol. 2013;200(4):373-383. https://doi.org/10.1083/jcb.201211138
PMid:23420871 PMCid:PMC3575529
- Costa Verdera H,
Gitz-Francois JJ, Schiffelers RM, Vader P. Cellular uptake of
extracellular vesicles is mediated by clathrin-independent endocytosis
and macropinocytosis. J Control Release. 2017;266:100-108.
https://doi.org/10.1016/j.jconrel.2017.09.019 PMid:28919558
- Mittelbrunn
M, Gutiérrez-Vázquez C, Villarroya-Beltri C, et al. Unidirectional
transfer of microRNA-loaded exosomes from T cells to antigen-presenting
cells. Nat Commun. 2011;2:282. https://doi.org/10.1038/ncomms1285
PMid:21505438 PMCid:PMC3104548
- Goloviznina NA, Verghese
SC, Yoon YM, Taratula O, Marks DL, Kurre P. Mesenchymal Stromal
Cell-derived Extracellular Vesicles Promote Myeloid-biased Multipotent
Hematopoietic Progenitor Expansion via Toll-Like Receptor Engagement. J
Biol Chem. 2016;291(47):24607- 24617.
https://doi.org/10.1074/jbc.M116.745653 PMid:27758863 PMCid:PMC5114412
- Crompot
E, Van Damme M, Pieters K, et al. Extracellular vesicles of bone marrow
stromal cells rescue chronic lymphocytic leukemia B cells from
apoptosis, enhance their migration and induce gene expression
modifications. Haematologica. 2017;102(9):1594-1604.
https://doi.org/10.3324/haematol.2016.163337 PMid:28596280
PMCid:PMC5685228
- Lin LY, Du LM, Cao K, et al. Tumour
cell-derived exosomes endow mesenchymal stromal cells with
tumour-promotion capabilities. Oncogene. 2016;35(46):6038-6042.
https://doi.org/10.1038/onc.2016.131 PMid:27132512 PMCid:PMC5116561
- Wolf
P. The nature and significance of platelet products in human plasma. Br
J Haematol. 1967;13(3):269-288.
https://doi.org/10.1111/j.1365-2141.1967.tb08741.x PMid:6025241
- Boilard
E, Duchez AC, Brisson A. The diversity of platelet microparticles. Curr
Opin Hematol. 2015;22(5):437-444.
https://doi.org/10.1097/MOH.0000000000000166 PMid:26214207
- Baj-Krzyworzeka
M, Majka M, Pratico D, et al. Platelet-derived microparticles stimulate
proliferation, survival, adhesion, and chemotaxis of hematopoietic
cells. Exp Hematol. 2002;30(5):450-459.
https://doi.org/10.1016/S0301-472X(02)00791-9
- Rozmyslowicz
T, Majka M, Kijowski J, et al. Platelet- and megakaryocyte-derived
microparticles transfer CXCR4 receptor to CXCR4-null cells and make
them susceptible to infection by X4-HIV. AIDS. 2003;17(1):33-42.
https://doi.org/10.1097/00002030-200301030-00006 PMid:12478067
- Ratajczak
MZ. Megakaryocyte-derived microvesicles, please stand up! Blood.
2009;113(5):981-982. https://doi.org/10.1182/blood-2008-10-182964
PMid:19179472
- Flaumenhaft R, Dilks JR, Richardson J, et
al. Megakaryocyte-derived microparticles: direct visualization and
distinction from platelet-derived microparticles. Blood.
2009;113(5):1112-1121. https://doi.org/10.1182/blood-2008-06-163832
PMid:18802008 PMCid:PMC2635076
- Jiang J, Kao CY,
Papoutsakis ET. How do megakaryocytic microparticles target and deliver
cargo to alter the fate of hematopoietic stem cells? J Control Release.
2017;247:1-18. https://doi.org/10.1016/j.jconrel.2016.12.021
PMid:28024915 PMCid:PMC5804484
- Nomura S, Nakamura T, Cone
J, Tandon NN, Kambayashi J. Cytometric analysis of high shear-induced
platelet microparticles and effect of cytokines on microparticle
generation. Cytometry. 2000;40(3):173-181.
https://doi.org/10.1002/1097-0320(20000701)40:3<173::AID-CYTO1>3.0.CO;2-L
- Vainchenker W, Kralovics R. Genetic basis and
molecular pathophysiology of classical myeloproliferative neoplasms.
Blood. 2017;129(6):667-679.
https://doi.org/10.1182/blood-2016-10-695940 PMid:28028029
- Desterke
C, Martinaud C, Ruzehaji N, Le Bousse-Kerdilès MC. Inflammation as a
Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis.
Mediators Inflamm. 2015;2015:415024.
https://doi.org/10.1155/2015/415024 PMid:26640324 PMCid:PMC4660030
- Tefferi
A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating
interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently
prognostic in primary myelofibrosis: a comprehensive cytokine profiling
study. J Clin Oncol. 2011;29(10):1356-1363.
https://doi.org/10.1200/JCO.2010.32.9490 PMid:21300928
- Hasselbalch
HC. Perspectives on chronic inflammation in essential thrombocythemia,
polycythemia vera, and myelofibrosis: is chronic inflammation a trigger
and driver of clonal evolution and development of accelerated
atherosclerosis and second cancer? Blood. 2012;119(14):3219-3225.
https://doi.org/10.1182/blood-2011-11-394775 PMid:22318201
- Al-Ali
HK, Griesshammer M, le Coutre P, et al. Safety and efficacy of
ruxolitinib in an open-label, multicenter, single-arm phase 3b
expanded-access study in patients with myelofibrosis: a snapshot of
1144 patients in the JUMP trial. Haematologica. 2016;101(9):1065-1073.
https://doi.org/10.3324/haematol.2016.143677 PMid:27247324
PMCid:PMC5060023
- Skoda RC, Duek A, Grisouard J.
Pathogenesis of myeloproliferative neoplasms. Exp Hematol.
2015;43(8):599-608. https://doi.org/10.1016/j.exphem.2015.06.007
PMid:26209551
- Zhan H, Ma Y, Lin CH, Kaushansky K. JAK2.
Leukemia. 2016;30(12):2332-2341. https://doi.org/10.1038/leu.2016.114
PMid:27133820 PMCid:PMC5158308
- Kuter DJ, Mufti GJ, Bain
BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow
reticulin formation in chronic immune thrombocytopenia patients treated
with romiplostim. Blood. 2009;114(18):3748-3756.
https://doi.org/10.1182/blood-2009-05-224766 PMid:19671919
- Villeval
JL, Cohen-Solal K, Tulliez M, et al. High thrombopoietin production by
hematopoietic cells induces a fatal myeloproliferative syndrome in
mice. Blood. 1997;90(11):4369- 4383. PMid:9373248
- Vannucchi
AM, Bianchi L, Cellai C, et al. Development of myelofibrosis in mice
genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood.
2002;100(4):1123-1132. https://doi.org/10.1182/blood-2002-06-1913
PMid:12149188
- Jantunen E, Hänninen A, Naukkarinen A,
Vornanen M, Lahtinen R. Gray platelet syndrome with splenomegaly and
signs of extramedullary hematopoiesis: a case report with review of the
literature. Am J Hematol. 1994;46(3):218-224.
https://doi.org/10.1002/ajh.2830460311 PMid:8192152
- Rameshwar
P, Narayanan R, Qian J, Denny TN, Colon C, Gascon P. NF-kappa B as a
central mediator in the induction of TGF-beta in monocytes from
patients with idiopathic myelofibrosis: an inflammatory response beyond
the realm of homeostasis. J Immunol. 2000;165(4):2271-2277.
https://doi.org/10.4049/jimmunol.165.4.2271 PMid:10925316
- Frey
BM, Rafii S, Teterson M, Eaton D, Crystal RG, Moore MA.
Adenovector-mediated expression of human thrombopoietin cDNA in
immune-compromised mice: insights into the pathophysiology of
osteomyelofibrosis. J Immunol. 1998;160(2):691-699. PMid:9551904
- Wagner-Ballon
O, Chagraoui H, Prina E, et al. Monocyte/macrophage dysfunctions do not
impair the promotion of myelofibrosis by high levels of thrombopoietin.
J Immunol. 2006;176(11):6425-6433.
https://doi.org/10.4049/jimmunol.176.11.6425 PMid:16709799
- Castro-Malaspina
H, Jhanwar SC. Properties of myelofibrosis-derived fibroblasts. Prog
Clin Biol Res. 1984;154:307-322. PMid:6382300
- Bowen-Pope
DF, Raines EW. History of discovery: platelet-derived growth factor.
Arterioscler Thromb Vasc Biol. 2011;31(11):2397-2401.
https://doi.org/10.1161/ATVBAHA.108.179556 PMid:22011752
PMCid:PMC3209478
- Bonner JC. Regulation of PDGF and its
receptors in fibrotic diseases. Cytokine Growth Factor Rev.
2004;15(4):255-273. https://doi.org/10.1016/j.cytogfr.2004.03.006
PMid:15207816
- Lucero HA, Ravid K, Grimsby JL, et al.
Lysyl oxidase oxidizes cell membrane proteins and enhances the
chemotactic response of vascular smooth muscle cells. J Biol Chem.
2008;283(35):24103-24117. https://doi.org/10.1074/jbc.M709897200
PMid:18586678 PMCid:PMC2527118
- Eliades A,
Papadantonakis N, Bhupatiraju A, et al. Control of megakaryocyte
expansion and bone marrow fibrosis by lysyl oxidase. J Biol Chem.
2011;286(31):27630-27638. https://doi.org/10.1074/jbc.M111.243113
PMid:21665949 PMCid:PMC3149354
- Papadantonakis N,
Matsuura S, Ravid K. Megakaryocyte pathology and bone marrow fibrosis:
the lysyl oxidase connection. Blood. 2012;120(9):1774-1781.
https://doi.org/10.1182/blood-2012-02-402594 PMid:22767499
PMCid:PMC3433087
- Gersuk GM, Carmel R, Pattengale PK.
Platelet-derived growth factor concentrations in platelet-poor plasma
and urine from patients with myeloproliferative disorders. Blood.
1989;74(7):2330-2334. PMid:2804368
- Lev PR, Marta RF,
Vassallu P, Molinas FC. Variation of PDGF, TGFbeta, and bFGF levels in
essential thrombocythemia patients treated with anagrelide. Am J
Hematol. 2002;70(2):85-91. https://doi.org/10.1002/ajh.10091
PMid:12111780
- Yoon SY, Tefferi A, Li CY. Cellular
distribution of platelet-derived growth factor, transforming growth
factor-beta, basic fibroblast growth factor, and their receptors in
normal bone marrow. Acta Haematol. 2000;104(4):151-157.
https://doi.org/10.1159/000046507 PMid:11279303
- Bock O,
Loch G, Büsche G, von Wasielewski R, Schlué J, Kreipe H. Aberrant
expression of platelet-derived growth factor (PDGF) and PDGF
receptor-alpha is associated with advanced bone marrow fibrosis in
idiopathic myelofibrosis. Haematologica. 2005;90(1):133-134.
PMid:15642683
- Bedekovics J, Kiss A, Beke L, Károlyi K,
Méhes G. Platelet derived growth factor receptor- beta (PDGFRβ)
expression is limited to activated stromal cells in the bone marrow and
shows a strong correlation with the grade of myelofibrosis. Virchows
Arch. 2013;463(1):57-65. https://doi.org/10.1007/s00428-013-1434-0
PMid:23748876
- Decker M, Martinez-Morentin L, Wang G, et
al. Leptin-receptor-expressing bone marrow stromal cells are
myofibroblasts in primary myelofibrosis. Nat Cell Biol.
2017;19(6):677-688. https://doi.org/10.1038/ncb3530 PMid:28481328
PMCid:PMC5801040
- Leask A, Abraham DJ. TGF-beta
signaling and the fibrotic response. FASEB J. 2004;18(7):816-827.
https://doi.org/10.1096/fj.03-1273rev PMid:15117886
- Martyré
MC, Magdelenat H, Bryckaert MC, Laine-Bidron C, Calvo F. Increased
intraplatelet levels of platelet-derived growth factor and transforming
growth factor-beta in patients with myelofibrosis with myeloid
metaplasia. Br J Haematol. 1991;77(1):80-86.
https://doi.org/10.1111/j.1365-2141.1991.tb07952.x PMid:1998600
- Le
Bousse-Kerdilès MC, Martyré MC. Dual implication of fibrogenic
cytokines in the pathogenesis of fibrosis and myeloproliferation in
myeloid metaplasia with myelofibrosis. Ann Hematol.
1999;78(10):437-444. https://doi.org/10.1007/s002770050595
PMid:10550553
- Wang JC, Novetsky A, Chen C, Novetsky AD.
Plasma matrix metalloproteinase and tissue inhibitor of
metalloproteinase in patients with agnogenic myeloid metaplasia or
idiopathic primary myelofibrosis. Br J Haematol. 2002;119(3):709-712.
https://doi.org/10.1046/j.1365-2141.2002.03874.x PMid:12437648
- Blank
U, Karlsson S. The role of Smad signaling in hematopoiesis and
translational hematology. Leukemia. 2011;25(9):1379-1388.
https://doi.org/10.1038/leu.2011.95 PMid:21566654
- Erba
BG, Gruppi C, Corada M, et al. Endothelial-to-Mesenchymal Transition in
Bone Marrow and Spleen of Primary Myelofibrosis. Am J Pathol.
2017;187(8):1879-1892. https://doi.org/10.1016/j.ajpath.2017.04.006
PMid:28728747
- Le Bousse-Kerdilès MC, Chevillard S,
Charpentier A, et al. Differential expression of transforming growth
factor-beta, basic fibroblast growth factor, and their receptors in
CD34+ hematopoietic progenitor cells from patients with myelofibrosis
and myeloid metaplasia. Blood. 1996;88(12):4534-4546. PMid:8977245
- Le
Bousse-Kerdilès MC, Martyré MC, Myelofibrosis FIrnoI. Involvement of
the fibrogenic cytokines, TGF-beta and bFGF, in the pathogenesis of
idiopathic myelofibrosis. Pathol Biol (Paris). 2001;49(2):153-157.
https://doi.org/10.1016/S0369-8114(00)00021-3
- Campanelli
R, Rosti V, Villani L, et al. Evaluation of the bioactive and total
transforming growth factor β1 levels in primary myelofibrosis.
Cytokine. 2011;53(1):100-106.
https://doi.org/10.1016/j.cyto.2010.07.427 PMid:20801055
- Ciaffoni
F, Cassella E, Varricchio L, Massa M, Barosi G, Migliaccio AR.
Activation of non- canonical TGF-β1 signaling indicates an autoimmune
mechanism for bone marrow fibrosis in primary myelofibrosis. Blood
Cells Mol Dis. 2015;54(3):234-241.
https://doi.org/10.1016/j.bcmd.2014.12.005 PMid:25703685
PMCid:PMC4338409
- Wernig G, Chen SY, Cui L, et al.
Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci
U S A. 2017;114(18):4757-4762. https://doi.org/10.1073/pnas.1621375114
PMid:28424250 PMCid:PMC5422830
- Vannucchi AM, Bianchi L,
Paoletti F, et al. A pathobiologic pathway linking thrombopoietin,
GATA-1, and TGF-beta1 in the development of myelofibrosis. Blood.
2005;105(9):3493-3501. https://doi.org/10.1182/blood-2004-04-1320
PMid:15665119
- Yanagida M, Ide Y, Imai A, et al. The
role of transforming growth factor-beta in PEG- rHuMGDF-induced
reversible myelofibrosis in rats. Br J Haematol. 1997;99(4):739-745.
https://doi.org/10.1046/j.1365-2141.1997.4843288.x PMid:9432016
- Chagraoui
H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F.
Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in
mice. Blood. 2002;100(10):3495- 3503.
https://doi.org/10.1182/blood-2002-04-1133 PMid:12393681
- Zingariello
M, Martelli F, Ciaffoni F, et al. Characterization of the TGF-β1
signaling abnormalities in the Gata1low mouse model of myelofibrosis.
Blood. 2013;121(17):3345-3363.
https://doi.org/10.1182/blood-2012-06-439661 PMid:23462118
PMCid:PMC3637011
- Ceglia I, Dueck AC, Masiello F, et al.
Preclinical rationale for TGF-β inhibition as a therapeutic target for
the treatment of myelofibrosis. Exp Hematol.
2016;44(12):1138-1155.e1134.
https://doi.org/10.1016/j.exphem.2016.08.007 PMid:27592389
PMCid:PMC5778911
- Gleissner CA, von Hundelshausen P, Ley
K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc
Biol. 2008;28(11):1920-1927. https://doi.org/10.1161/ATVBAHA.108.169417
PMid:18723831 PMCid:PMC2657037
- Lasagni L, Francalanci
M, Annunziato F, et al. An alternatively spliced variant of CXCR3
mediates the inhibition of endothelial cell growth induced by IP-10,
Mig, and I-TAC, and acts as functional receptor for platelet factor 4.
J Exp Med. 2003;197(11):1537-1549. https://doi.org/10.1084/jem.20021897
PMid:12782716 PMCid:PMC2193908
- Zaldivar MM, Pauels K,
von Hundelshausen P, et al. CXC chemokine ligand 4 (Cxcl4) is a
platelet-derived mediator of experimental liver fibrosis. Hepatology.
2010;51(4):1345-1353. https://doi.org/10.1002/hep.23435 PMid:20162727
- van
Bon L, Affandi AJ, Broen J, et al. Proteome-wide analysis and CXCL4 as
a biomarker in systemic sclerosis. N Engl J Med. 2014;370(5):433-443.
https://doi.org/10.1056/NEJMoa1114576 PMid:24350901 PMCid:PMC4040466
- Burstein
SA, Malpass TW, Yee E, et al. Platelet factor-4 excretion in
myeloproliferative disease: implications for the aetiology of
myelofibrosis. Br J Haematol. 1984;57(3):383-392.
https://doi.org/10.1111/j.1365-2141.1984.tb02912.x PMid:6743563
- Schneider
RK, Mullally A, Dugourd A, et al. Gli1. Cell Stem Cell. 2017;20(6):785-
800.e788. https://doi.org/10.1016/j.stem.2017.03.008 PMid:28457748
- Gómez-Lechón
MJ. Oncostatin M: signal transduction and biological activity. Life
Sci. 1999;65(20):2019-2030.
https://doi.org/10.1016/S0024-3205(99)00296-9
- Mosley B,
De Imus C, Friend D, et al. Dual oncostatin M (OSM) receptors. Cloning
and characterization of an alternative signaling subunit conferring
OSM-specific receptor activation. J Biol Chem.
1996;271(51):32635-32643. https://doi.org/10.1074/jbc.271.51.32635
PMid:8999038
- Tanaka M, Hirabayashi Y, Sekiguchi T,
Inoue T, Katsuki M, Miyajima A. Targeted disruption of oncostatin M
receptor results in altered hematopoiesis. Blood. 2003;102(9):3154-
3162. https://doi.org/10.1182/blood-2003-02-0367 PMid:12855584
- Minehata
K, Takeuchi M, Hirabayashi Y, et al. Oncostatin m maintains the
hematopoietic microenvironment and retains hematopoietic progenitors in
the bone marrow. Int J Hematol. 2006;84(4):319-327.
https://doi.org/10.1532/IJH97.06090 PMid:17118758
- Hoermann
G, Cerny-Reiterer S, Perné A, et al. Identification of oncostatin M as
a STAT5- dependent mediator of bone marrow remodeling in KIT
D816V-positive systemic mastocytosis. Am J Pathol.
2011;178(5):2344-2356. https://doi.org/10.1016/j.ajpath.2011.01.020
PMid:21457934 PMCid:PMC3081146
- Wallace PM, MacMaster
JF, Rillema JR, Peng J, Burstein SA, Shoyab M. Thrombocytopoietic
properties of oncostatin M. Blood. 1995;86(4):1310-1315 PMid:7632937
- Hoermann
G, Cerny-Reiterer S, Herrmann H, et al. Identification of oncostatin M
as a JAK2 V617F-dependent amplifier of cytokine production and bone
marrow remodeling in myeloproliferative neoplasms. FASEB J.
2012;26(2):894-906. https://doi.org/10.1096/fj.11-193078 PMid:22051730
- Schepers
K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels
the endosteal bone marrow niche into a self-reinforcing leukemic niche.
Cell Stem Cell. 2013;13(3):285-299.
https://doi.org/10.1016/j.stem.2013.06.009 PMid:23850243
PMCid:PMC3769504
- Murphy PM. Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin Hematol. 1997;34(4):311-318. PMid:9347581
- Takeuchi
K, Higuchi T, Yamashita T, Koike K. Chemokine production by human
megakaryocytes derived from CD34-positive cord blood cells. Cytokine.
1999;11(6):424-434. https://doi.org/10.1006/cyto.1998.0455
PMid:10346982
- Emadi S, Clay D, Desterke C, et al. IL-8
and its CXCR1 and CXCR2 receptors participate in the control of
megakaryocytic proliferation, differentiation, and ploidy in myeloid
metaplasia with myelofibrosis. Blood. 2005;105(2):464-473.m
https://doi.org/10.1182/blood-2003-12-4415 PMid:15454487
- Hol
J, Wilhelmsen L, Haraldsen G. The murine IL-8 homologues KC, MIP-2, and
LIX are found in endothelial cytoplasmic granules but not in
Weibel-Palade bodies. J Leukoc Biol. 2010;87(3):501-508.
https://doi.org/10.1189/jlb.0809532 PMid:20007247
- Bock
O, Höftmann J, Theophile K, et al. Bone morphogenetic proteins are
overexpressed in the bone marrow of primary myelofibrosis and are
apparently induced by fibrogenic cytokines. Am J Pathol.
2008;172(4):951-960. https://doi.org/10.2353/ajpath.2008.071030
PMid:18349123 PMCid:PMC2276425
- Garimella R, Kacena MA,
Tague SE, Wang J, Horowitz MC, Anderson HC. Expression of bone
morphogenetic proteins and their receptors in the bone marrow
megakaryocytes of GATA- 1(low) mice: a possible role in osteosclerosis.
J Histochem Cytochem. 2007;55(7):745-752.
https://doi.org/10.1369/jhc.6A7164.2007 PMid:17371937
- Lu
M, Xia L, Liu YC, et al. Lipocalin produced by myelofibrosis cells
affects the fate of both hematopoietic and marrow microenvironmental
cells. Blood. 2015;126(8):972-982.
https://doi.org/10.1182/blood-2014-12-618595 PMid:26022238
PMCid:PMC4543230
- Martyré MC, Le Bousse-Kerdiles MC,
Romquin N, et al. Elevated levels of basic fibroblast growth factor in
megakaryocytes and platelets from patients with idiopathic
myelofibrosis. Br J Haematol. 1997;97(2):441-448.
https://doi.org/10.1046/j.1365-2141.1997.292671.x PMid:9163611
- Boiocchi
L, Vener C, Savi F, et al. Increased expression of vascular endothelial
growth factor receptor 1 correlates with VEGF and microvessel density
in Philadelphia chromosome- negative myeloproliferative neoplasms. J
Clin Pathol. 2011;64(3):226-231.
https://doi.org/10.1136/jcp.2010.083386 PMid:21217153
- Ruberti
S, Bianchi E, Guglielmelli P, et al. Involvement of MAF/SPP1 axis in
the development of bone marrow fibrosis in PMF patients. Leukemia.
2018;32(2):438-449. https://doi.org/10.1038/leu.2017.220 PMid:28745329
PMCid:PMC5808097
- Evrard S, Bluteau O, Tulliez M, et al.
Thrombospondin-1 is not the major activator of TGF- β1 in
thrombopoietin-induced myelofibrosis. Blood. 2011;117(1):246-249.
https://doi.org/10.1182/blood-2010-07-294447 PMid:20944070
- Muth
M, Engelhardt BM, Kröger N, et al. Thrombospondin-1 (TSP-1) in primary
myelofibrosis (PMF) - a megakaryocyte-derived biomarker which largely
discriminates PMF from essential thrombocythemia. Ann Hematol.
2011;90(1):33-40. https://doi.org/10.1007/s00277-010-1024-z
PMid:20625903
- Abbonante V, Di Buduo CA, Gruppi C, et
al. A new path to platelet production through matrix sensing.
Haematologica. 2017. https://doi.org/10.3324/haematol.2016.161562
PMCid:PMC5566016
- Malara A, Currao M, Gruppi C, et al.
Megakaryocytes contribute to the bone marrow-matrix environment by
expressing fibronectin, type IV collagen, and laminin. Stem Cells.
2014;32(4):926- 937. https://doi.org/10.1002/stem.1626 PMid:24357118
PMCid:PMC4096110
- Abbonante V, Di Buduo CA, Gruppi C, et
al. Thrombopoietin/TGF-β1 Loop Regulates Megakaryocyte Extracellular
Matrix Component Synthesis. Stem Cells. 2016;34(4):1123-1133.
https://doi.org/10.1002/stem.2285 PMid:26748484
- Malara
A, Gruppi C, Rebuzzini P, et al. Megakaryocyte-matrix interaction
within bone marrow: new roles for fibronectin and factor XIII-A. Blood.
2011;117(8):2476-2483. https://doi.org/10.1182/blood-2010-06-288795
PMid:21131589
- Malara A, Gruppi C, Rebuzzini P, et al.
Megakaryocyte-matrix interaction within bone marrow: New roles for
fibronectin and factor XIII-A. Blood. 2011;117(8):2476-2483.
https://doi.org/10.1182/blood-2010-06-288795 PMid:21131589
- Mosher
DF, Schad PE, Vann JM. Cross-linking of collagen and fibronectin by
factor XIIIa. Localization of participating glutaminyl residues to a
tryptic fragment of fibronectin. J Biol Chem. 1980;255(3):1181-1188.
PMid:7356656
- Abbonante V, Chitalia V, Rosti V, et al.
Upregulation of lysyl oxidase and adhesion to collagen of human
megakaryocytes and platelets in primary myelofibrosis. Blood.
2017;130(6):829- 831. https://doi.org/10.1182/blood-2017-04-777417
PMid:28592432 PMCid:PMC5553577
- Rundhaug JE. Matrix
metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9(2):267-
285. https://doi.org/10.1111/j.1582-4934.2005.tb00355.x PMid:15963249
- Lane
WJ, Dias S, Hattori K, et al. Stromal-derived factor 1-induced
megakaryocyte migration and platelet production is dependent on matrix
metalloproteinases. Blood. 2000;96(13):4152-4159. PMid:11110686
- Villeneuve
J, Block A, Le Bousse-Kerdilès MC, et al. Tissue inhibitors of matrix
metalloproteinases in platelets and megakaryocytes: a novel
organization for these secreted proteins. Exp Hematol.
2009;37(7):849-856. https://doi.org/10.1016/j.exphem.2009.03.009
PMid:19410025
- Wang JC. Importance of plasma matrix
metalloproteinases (MMP) and tissue inhibitors of metalloproteinase
(TIMP) in development of fibrosis in agnogenic myeloid metaplasia. Leuk
Lymphoma. 2005;46(9):1261-1268.
https://doi.org/10.1080/10428190500126463 PMid:16109602
- Bock
O, Neuse J, Hussein K, et al. Aberrant collagenase expression in
chronic idiopathic myelofibrosis is related to the stage of disease but
not to the JAK2 mutation status. Am J Pathol.
2006;169(2):471-481.https://doi.org/10.2353/ajpath.2006.060110
PMid:16877349 PMCid:PMC1780160
- Wickenhauser C, Schmitz
B, Baldus SE, et al. Selectins (CD62L, CD62P) and megakaryocytic
glycoproteins (CD41a, CD42b) mediate megakaryocyte-fibroblast
interactions in human bone marrow. Leuk Res. 2000;24(12):1013-1021.
https://doi.org/10.1016/S0145-2126(00)00063-1
- Abbonante
V, Gruppi C, Catarsi P, et al. Altered fibronectin expression and
deposition by myeloproliferative neoplasm-derived mesenchymal stromal
cells. Br J Haematol. 2016;172(1):140- 144.
https://doi.org/10.1111/bjh.13471 PMid:25940987
- Avanzini
MA, Abbonante V, Catarsi P, et al. The spleen of patients with
myelofibrosis harbors defective mesenchymal stromal cells. Am J
Hematol. 2018. https://doi.org/10.1002/ajh.25047 PMid:29359451
- Schneider
RK, Ziegler S, Leisten I, et al. Activated fibronectin-secretory
phenotype of mesenchymal stromal cells in pre-fibrotic
myeloproliferative neoplasms. J Hematol Oncol. 2014;7:92.
https://doi.org/10.1186/s13045-014-0092-2 PMid:25498831
PMCid:PMC4271470
- Cashell AW, Buss DH. The frequency and
significance of megakaryocytic emperipolesis in myeloproliferative and
reactive states. Ann Hematol. 1992;64(6):273-276.
https://doi.org/10.1007/BF01695470
- Schmitt A,
Drouin A, Massé JM, Guichard J, Shagraoui H, Cramer EM.
Polymorphonuclear neutrophil and megakaryocyte mutual involvement in
myelofibrosis pathogenesis. Leuk Lymphoma. 2002;43(4):719-724.
https://doi.org/10.1080/10428190290016809 PMid:12153156
- Centurione
L, Di Baldassarre A, Zingariello M, et al. Increased and pathologic
emperipolesis of neutrophils within megakaryocytes associated with
marrow fibrosis in GATA- 1(low) mice. Blood. 2004;104(12):3573-3580.
https://doi.org/10.1182/blood-2004-01-0193 PMid:15292068
- Zetterberg
E, Verrucci M, Martelli F, et al. Abnormal P-selectin localization
during megakaryocyte development determines thrombosis in the gata1low
model of myelofibrosis. Platelets. 2014;25(7):539-547.
https://doi.org/10.3109/09537104.2013.840720 PMid:24176039
PMCid:PMC4045657
- Spangrude GJ, Lewandowski D, Martelli
F, et al. P-Selectin Sustains Extramedullary Hematopoiesis in the Gata1
low Model of Myelofibrosis. Stem Cells. 2016;34(1):67-82.
https://doi.org/10.1002/stem.2229 PMid:26439305
- Di
Buduo CA, Alberelli MA, Glembostky AC, et al. Abnormal proplatelet
formation and emperipolesis in cultured human megakaryocytes from gray
platelet syndrome patients. Sci Rep. 2016;6:23213.
https://doi.org/10.1038/srep23213 PMid:26987485 PMCid:PMC4796794
- Ghafoory
S, Varshney R, Robison T, et al. Platelet TGF-β1 deficiency decreases
liver fibrosis in a mouse model of liver injury. Blood Adv.
2018;2(5):470-480. https://doi.org/10.1182/bloodadvances.2017010868
PMid:29490978 PMCid:PMC5851416
- Kodama T, Takehara T,
Hikita H, et al. Thrombocytopenia exacerbates cholestasis-induced liver
fibrosis in mice. Gastroenterology. 2010;138(7):2487-2498,
2498.e2481-2487.
- Meyer A, Wang W, Qu J, et al. Platelet
TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic
dysfunction in a mouse model of pressure overload. Blood.
2012;119(4):1064- 1074. https://doi.org/10.1182/blood-2011-09-377648
PMid:22134166 PMCid:PMC3271718
- Piguet PF, Vesin C.
Pulmonary platelet trapping induced by bleomycin: correlation with
fibrosis and involvement of the beta 2 integrins. Int J Exp Pathol.
1994;75(5):321-328. PMid:7528044 PMCid:PMC2001873
- Guo
Y, Mishra A, Howland E, et al. Platelet-derived Wnt antagonist
Dickkopf-1 is implicated in ICAM-1/VCAM-1-mediated neutrophilic acute
lung inflammation. Blood. 2015;126(19):2220-2229.
https://doi.org/10.1182/blood-2015-02-622233 PMid:26351298
PMCid:PMC4635118
- Rondina MT, Weyrich AS. Regulation of
the genetic code in megakaryocytes and platelets. J Thromb Haemost.
2015;13 Suppl 1:S26-32. https://doi.org/10.1111/jth.12965 PMid:26149034
PMCid:PMC4498409
- Freishtat RJ, Natale J, Benton AS, et
al. Sepsis alters the megakaryocyte-platelet transcriptional axis
resulting in granzyme B-mediated lymphotoxicity. Am J Respir Crit Care
Med. 2009;179(6):467-473. https://doi.org/10.1164/rccm.200807-1085OC
PMid:19136373 PMCid:PMC2654976
- Zaslavsky A, Baek KH,
Lynch RC, et al. Platelet-derived thrombospondin-1 is a critical
negative regulator and potential biomarker of angiogenesis. Blood.
2010;115(22):4605-4613. https://doi.org/10.1182/blood-2009-09-242065
PMid:20086246 PMCid:PMC2881490
- Wen QJ, Yang Q,
Goldenson B, et al. Targeting megakaryocytic-induced fibrosis in
myeloproliferative neoplasms by AURKA inhibition. Nat Med.
2015;21(12):1473-1480. https://doi.org/10.1038/nm.3995 PMid:26569382
PMCid:PMC4674320
[TOP]