Alessandro Matte1, Francesco Zorzi1, Filippo Mazzi1, Enrica Federti1, Oliviero Olivieri1 and Lucia De Franceschi1.
1 Department of Medicine, University of Verona and AOUI Verona, Verona. Italy
Correspondence to: Lucia De Franceschi. Department of Medicine, University of
Verona and AOUI Verona, Verona; Italy. Fax: +39 045 8027473; phone:
+39045 8124401. E-mail:
lucia.defranceschi@univr.it
Published: January 1, 2019
Received: October 1, 2018
Accepted: November 11, 2018
Mediterr J Hematol Infect Dis 2019, 11(1): e2019002 DOI
10.4084/MJHID.2019.002
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Sickle
cell disease (SCD; ORPHA232; OMIM # 603903) is a chronic and
invalidating disorder distributed worldwide, with high morbidity and
mortality. Given the disease complexity and the multiplicity of
pathophysiological targets, development of new therapeutic options is
critical, despite the positive effects of hydroxyurea (HU), for many
years the only approved drug for SCD.
New therapeutic strategies might be divided into (1) pathophysiology-related novel therapies and
(2)
innovations in curative therapeutic options such as hematopoietic stem
cell transplantation and gene therapy. The pathophysiology related
novel therapies are: a) Agents which reduce sickling or prevent sickle
red cell dehydration; b) Agents targeting SCD vasculopathy and sickle
cell- endothelial adhesive events; c) Anti-oxidant agents.
This
review highlights new therapeutic strategies in SCD and discusses
future developments, research implications, and possible innovative
clinical trials.
|
Introduction
Sickle
cell disease (SCD) is a hemoglobinopathy which affects approximately
100,000 individuals in the United States and almost 20,000-25,000
subjects in Europe, mainly immigrants from endemic areas such as
Sub-Saharan Africa to European countries.[1-3] Estimates of the number of affected newborn in 2010 are of approximately 312,302 subjects with 75.5% being born in Africa.[4] The invalidating impact of SCD on patient survival, quality of life and cost for health systems,[2] requires the development of new therapeutic options to treat sickle cell related acute and chronic complications.
SCD
is caused by a point mutation in the β-globin gene resulting in the
synthesis of pathological hemoglobin S (HbS). HbS displays peculiar
biochemical characteristics, polymerizing when deoxygenated with
associated reduction in cell ion and water content (cell dehydration),
increased red cell density and further acceleration of HbS
polymerization (Figure 1).[5-7]
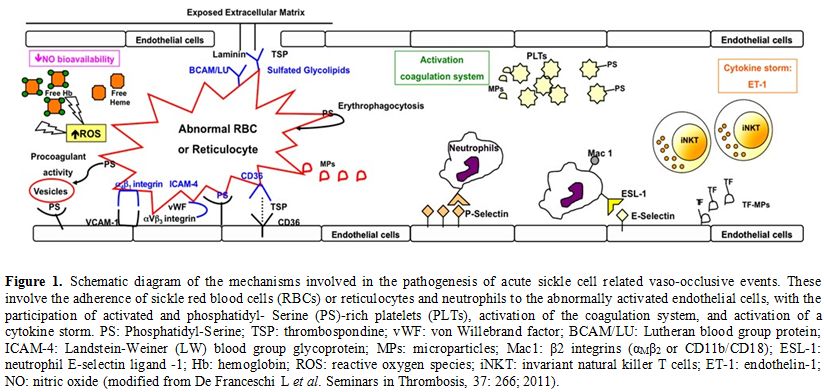 |
Figure 1. Schematic diagram of the mechanisms involved in the pathogenesis of
acute sickle cell related vaso-occlusive events. These involve the
adherence of sickle red blood cells (RBCs) or reticulocytes and
neutrophils to the abnormally activated endothelial cells, with the
participation of activated and phosphatidyl- Serine (PS)-rich platelets
(PLTs), activation of the coagulation system, and activation of a
cytokine storm. PS: Phosphatidyl-Serine; TSP: thrombospondine; vWF: von
Willebrand factor; BCAM/LU: Lutheran blood group protein; ICAM-4:
Landstein-Weiner (LW) blood group glycoprotein; MPs: microparticles;
Mac1: β2 integrins (αMβ2 or CD11b/CD18); ESL-1: neutrophil E- selectin
ligand -1; Hb: hemoglobin; ROS: reactive oxygen species; iNKT:
invariant natural killer T cells; ET-1: endothelin-1; NO: nitric oxide
(modified from De Franceschi L et al. Seminars in Thrombosis, 37: 266;
2011). |
Pathophysiological
studies have shown that dense, dehydrated red cells play a central role
in acute and chronic clinical manifestations of SCD, in which
intravascular sickling in capillaries and small vessels leads to
vaso-occlusion and impaired blood flow with ischemic/reperfusion
injury.[5,8-10] In microcirculation,
vaso-occlusive events (VOC) result from a complex and still partially
known scenario, involving the interactions between different cell
types, including dense red cells, reticulocytes, abnormally activated
endothelial cells, leukocytes, platelets and plasma factors (Figure 1).[5,9-13]
Acute VOCs have been associated with increased expression of
pro-adhesion molecules such as vascular adhesion molecule-1 (VCAM-1),
intracellular adhesion molecule-1 (ICAM-1) or selectins (Figure 1).[5,9,11,12,14,15]
These molecules are important in recruitment and adhesion of both
neutrophils and sickle red cells to the abnormally activated vascular
endothelial surface.[11,16] In
addition, the presence of free Hb and free heme contribute to the local
reduction of nitric oxide (NO) bioavailability, establishing an
endovascular high pro- oxidant and pro-inflammatory environment. This
is associated with modulation of innate immunity and increased iNKT
lymphocytes, increase levels of vascular active cytokines such as
endothelin 1, combined with the final contribution of platelets (Figure 1).[5,9,14,17-20] Hydroxyurea is the Gold- Standard Treatment for Sickle Cell Disease
Hydroxyurea
or hydroxycarbamide (HU) is the key therapeutic tool for SCD approved
by Food and Drug Administration (FDA) and European Medical Agency
(EMEA). US and European guidelines highlighted that HU should be
available for all SCD patients from pediatric to adult populations.[21,22]
Studies
in SCD show a multimodal action of HU, which (i) increases HbF
production, resulting in delayed HbS polymerization; (ii) reduces
hemolysis and increase NO availability targeting cGMP production; (iii)
modulates endothelial activation and reduces neutrophil counts,
contributing to the reduction of chronic inflammation (Figure 2).[23-27]
Long-term use of HU has been shown to be safe and well-tolerated in
large cohorts of children and adults with SCD, reducing mortality and
morbidity of both children and adult patients.[21,28-31]
Indeed, HU reduces (i) the frequency of VOC and the rate of
hospitalization; (ii) the incidence of ACS; (iii) the transfusion
requirements; and (iv) the severity of dactilitis in SCD pediatric
population.[21,32-36] HU might also
be used in combination with transfusion regimen in selected SCD
population such as SCD children with progressive cerebrovascular
disease in the absence of antigen- matched sibling donor.[37]
Furthermore, recent reports propose HU as acceptable alternative to
chronic transfusion regimen in SCD patients with history of
abnormalities at the transcranial doppler scan (TCD), used to screen
for cerebrovascular disease in pediatric patients.[38-40]
This requires a close follow-up by TCD scan every 3 months, with the
possibility to switch-back to chronic transfusion regimen if abnormal
transcranial velocities are again documented.[38-40]
Noteworthy, increase reticulocyte count before HU treatment and high
leukocyte count after HU have been identified as risk factor for
reversion to abnormal TCD velocities in SCD pediatric patients. Thus,
again chronic inflammation and vasculopathy seems to be key
determinants of severe chronic complications in SCD.[38-40]
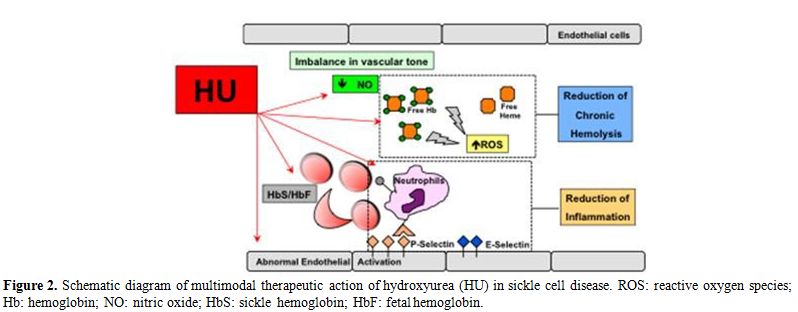 |
Figure 2. Schematic
diagram of multimodal therapeutic action of hydroxyurea (HU) in sickle
cell disease. ROS: reactive oxygen species; Hb: hemoglobin; NO: nitric
oxide; HbS: sickle hemoglobin; HbF: fetal hemoglobin. |
Although
HU should be available for all SCD subjects, the major limitation is
the poor adherence of adults SCD patients to HU therapy. Different
studies have identified multiple factors to be involved in reduced
adherence of SCD patients to HU such as (i) chronicity of the
treatment; (ii) socio-economic reasons; and (iii) adhesion barriers
related to the transition from pediatric to adult care system.[41-44]
The
dissemination of the use of HU is particularly important in
underdeveloped countries with high incidence of SCD such as in the
sub-Saharan African areas.[45] Recently, Opoka et al.
reported safety of use for HU at the dosage of ~20 mg/Kg/d in African
children from Uganda, a malaria endemic area (NOHARM study,
NCT01976416).[46] This study further supports the
importance of HU as a front-line medical treatment for SCD patients all
over the world. Noteworthy, in geographical context where frequent
hematologic monitoring is not available, Toya et al. have recently
reported the beneficial effects of low dose HU (10 mg/Kg/d) on SCD
acute clinical manifestations in Nigerian patients.[47]
Novel Therapeutic Approaches to Treat Sickle Cell Disease
In
the last two decades, the availability of mouse models for SCD has
allowed both characterization of the pathogenesis of sickle cell
related organ damage(s) and identification of pathophysiology- based
new therapeutic options in addition to HU.[5,7,11,12,48-50]
As shown in Table 1, pathophysiology related novel therapies for SCD can be divided into:
• Agents which reduce/prevent sickle red cell dehydration or red cell sickling or HbF inducers;
• Agents targeting SCD vasculopathy and sickle cell- endothelial adhesive events;
• Anti-oxidant agents.
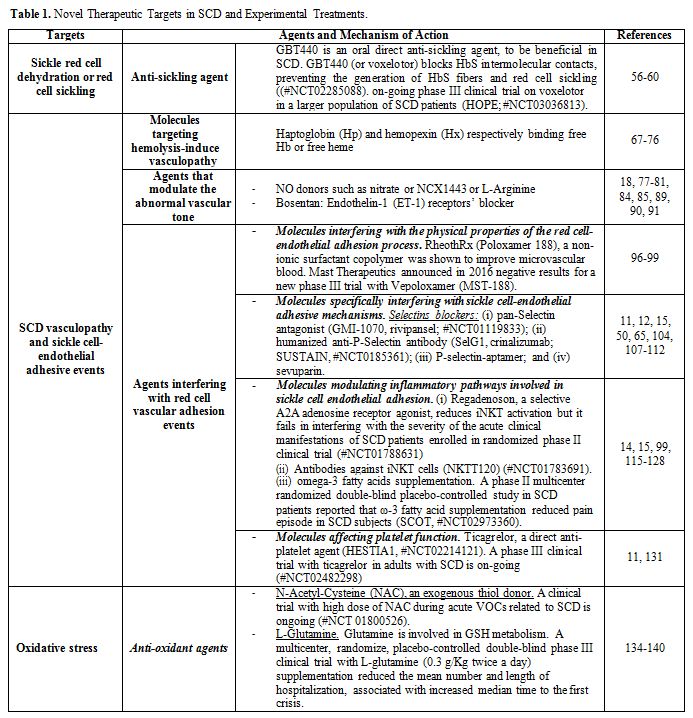 |
Table 1. Targets in SCD and Experimental Treatments. |
Agents Which Reduce/Prevent Sickle Red Cell Dehydration and Sickling.
Different agents targeting sickle red cells have been developed to
prevent or limit HbS polymerization or to block the mechanism(s)
involved in red cell dehydration.[14,18,19,48,51-55]
Targeting the reduction of circulating dense red cells and/or sickled
red cells is very important, since these cells are easily trapped in
microcirculation and participate to the pathogenesis of acute VOC.
Recent
reports indicate GBT440, an oral direct anti-sickling agent, to be
beneficial in SCD. GBT440 (or voxelotor) blocks HbS intermolecular
contacts, preventing the generation of HbS fibers and red cell
sickling.[56-60] GBT440 has been shown (i) to
ameliorate in vitro SCD red cell features such as red cell
deformability or viscosity and (ii) to improve sickle red cell survival
with decrease reticulocyte count.[56-60] Preliminary
data on phase I/II double blind placebo study with GBT440 in healthy
volunteers and few SCD patients show safety and tolerability of
GBT440 associated with an amelioration of hemolytic indices and a
reduction in reticulocyte count (#NCT02285088).[55,61,62]
Blyden et al. have reported the compassionate use of voxelotor, at the
dosage of 900 mg/d up to 1500 mg/d for 24 weeks in a small group of
subjects with severe untreatable SCD. Voxelotor beneficially impacts
SCD patient well-being with a reduction in number of hospitalization
for severe VOC compared to patient’s clinical history.[63]
These data further support the on-going phase III clinical trial on
voxelotor in a larger population of SCD patients (HOPE; #NCT03036813).
Agents Targeting SCD Vasculopathy and Sickle Cell-Endothelial Adhesive Events.
SCD is not only a hemolytic anemia but also a chronic inflammatory
disorder characterized by abnormally activated vascular endothelial
cells, amplified inflammatory response, and the release of soluble
factors, which promote abnormal adhesive interactions between
erythrocytes, endothelial cells, and neutrophils.[5,7,10,12,64,65]
An increased number of circulating, abnormally activated endothelial
cells has been identified in SCD patients during acute VOCs, indicating
the presence of chronic vasculopathy, worsened by acute events.[66]
Thus, SCD is characterized by a chronic inflammatory vasculopathy that
favors the recruitment of leukocytes and the entrapment of dense red
cells with the generation of heterotypic aggregates (thrombi) with
ischemic/reperfusion local damage.
In this context, the major
objectives of therapeutic strategies targeting sickle cell vasculopathy
are to reduce or prevent vascular endothelial activation and damage.
The end-point of anti-adherence therapy, alternatively, is to interfere
with the initialization and/or amplification of adhesive events.
In SCD, agents targeting SCD vasculopathy and sickle cell-endothelial adhesive events (Figure 3) can be divided into:
i. Molecules targeting hemolysis-induced vasculopathy;
ii. Agents that modulate the abnormal vascular tone;
iii. Agents interfering with red cell vascular adhesion events.
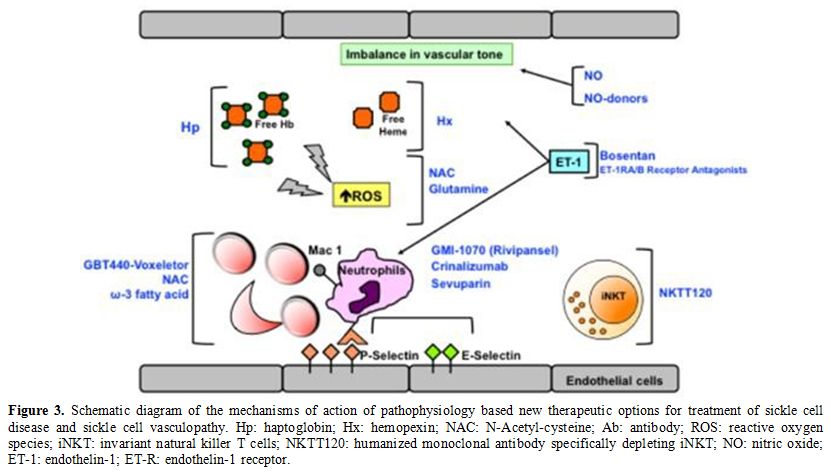 |
Figure 3. Schematic
diagram of the mechanisms of action of pathophysiology based new
therapeutic options for treatment of sickle cell disease and sickle
cell vasculopathy. Hp: haptoglobin; Hx: hemopexin; NAC:
N-Acetyl-cysteine; Ab: antibody; ROS: reactive oxygen species; iNKT:
invariant natural killer T cells; NKTT120: humanized monoclonal
antibody specifically depleting iNKT; NO: nitric oxide; ET-1:
endothelin- 1; ET-R: endothelin-1 receptor. |
i. Molecules targeting hemolysis-induced vasculopathy.
The chronic hemolytic anemia of SCD is
for one-third intravascular and for two-third
extravascular, via the reticulo-endothelial systems. Free Hb is present
in the peripheral circulation of SCD patients, reacting with plasma
nitric oxide (NO) with production of reactive oxygen species (ROS) and
generation of MetHb. This is a key step for the release of free heme.[9,67,68]
The physiological systems binding free Hb or free heme are haptoglobin (Hp) and hemopexin (Hx), respectively.
In
SCD patients, both Hp and Hx levels are significantly reduced in steady
state compared to healthy controls; they further decrease during acute
VOCs.[67,69] The highly pro-oxidant
environment with the presence of free heme and free Hb promotes
inflammation and abnormal vascular activation with increased expression
of adhesion vascular molecules such as VCAM-1, ICAM-1 or E-selectin.[67,69]
Studies in mouse models for SCD have shown that free heme induces
vascular stasis and leukocyte extravasation with the trapping of dense
red cells and neutrophils in microcirculation.[70-72]
In human SCD patients, free Hb and free heme increase during acute VOCs with further reduction in Hp and Hx levels (Figure 1).[72,73] Noteworthy, Hp levels correlate with pulmonary hypertension,[67]
suggesting that the blockage of free-Hb by Hp might possibly affect SCD
related organ damage. In mouse models for SCD, the infusion of Hp has
been shown to prevent vascular stasis. Encouraging data from small, in
vivo human studies with infused Hp show that Hp protects the kidneys
from free Hb-related tubular damage in patients who have undergone
cardiopulmonary surgery or endoscopic sclerotherapy.[67]
Few case reports are present in the literature on the use of Hp in
patients with hemolytic crisis and inherited red cell disorders.[67,74] Thus, Hp might be as a possible new therapeutic tool to be further explored in SCD.
In
the complex scenario of the pathogenesis of SCD vasculopathy, Hx, a
high affinity heme binding protein, represents another interesting
molecule that might be explored as a novel therapeutic option (Figure 1).
The supplementation of Hx in mouse models for SCD has been shown to
reduce heme induced oxidative stress, vascular endothelial injury,
inflammation, and vascular stasis.[9] Recently, a link
between increased free heme and complement activation has been reported
in cell- and animal-based model for SCD.[75] Hx
significantly reduces complement deposition in kidney from humanized
SCD mice, highlighting the importance of controlling free heme plasma
level as additional tool to limit inflammatory vasculopathy and related
severe organ damage in SCD. The importance of optimal levels of Hp and
Hx is also supported by a recent report on the use of therapeutic
plasma exchange in SCD with severe VOC, resistant to red cell exchange.[76]
Further
studies need be carried out to develop and understand the potential
clinical use of Hp and/or Hx in management of severe complication
related to excess of free heme in SCD patients.
ii. Agents that modulate the abnormal vascular tone.
Vascular tone results from the balance between vaso-dilatatory factors
such as nitric oxide (NO) and vaso-constrictor factors such as the
endothelin-1 (ET-1) system (Figure 1).[18,77-81]
In SCD, reduced NO local bioavailability, a consequence of the presence
of free Hb, contributes to chronic vaso-constriction and amplifies the
expression of vascular adhesion molecules.[77,82,83]
In addition, the release of arginase in peripheral circulation by
sickle red cells during chronic hemolysis, subtracts arginine from the
urea cycle in endothelial cells, and further contributes to NO
deficiency.[77,82-85] Plasma NO
metabolites are decreased in SCD patients during acute VOCs and
decreased exhaled NO has also been reported. Thus, therapeutic
strategies to supplement or modulate NO might beneficially interfere
with the pathogenesis of acute SCD related clinical manifestations such
as VOCs. Initial trials showed some positive, encouraging effects of
inhaled NO on acute VOCs.[82,86,87]
However, a subsequent multicentric, double-blind, randomized
placebo-controlled study in SCD with VOCs using inhaled NO showed no
clinically significant effects.[82] New NO donors such as nitrate or NCX1443 need to be further evaluated in humanized animal-based pre- clinical studies.[83,88]
Another possible strategy to increase NO production in SCD is the
supplementation of L- Arginine. Oral L-Arginine (i) decreases artery
pulmonary pressure in SCD; (ii) improves leg ulcers; and (iii)
contributes in pain control in SCD.[84,85,89]
The co-administration of L-Arginine with HU has been reported to
increase levels of nitrate, suggesting L- Arginine as an adjuvant
molecule in treatment of SCD.[84,85,89]
Endothelin-1
is a potent vasoconstrictor and bronchoconstrictor, whose plasma and
urinary values are increased in SCD subjects in steady state and during
acute VOCs.[18,90,91] In a mouse
model for SCD, the ET-1 receptors’ blocker, bosentan, prevented hypoxia
induced organ damage and affect neutrophil mediated inflammatory
response, suggesting the modulation of the ET-1 system as an additional
therapeutic option to interfere with the pathogenesis of SCD related
clinical manifestation(s).[18,92,93]
It is of interest to note that increased ET-1 and high ET-1 levels have
been shown to positively correlate with pain rating in children with
SCD.[94] This has been recently investigated in
humanized mouse model for SCD, showing that endothelin receptor-type A
might be involved in inflammatory mediated pain component throughout
the modulation of Nav1.8 channel in primary sensing neurons.[95]
iii. Agents interfering with red cell vascular adhesion events.
In SCD, anti-adherence therapeutic strategies might represent an
interesting, novel therapeutic strategy to prevent the generation of
acute VOCs and to lessen SCD related organ damage (Figure 1 and 3). The anti-adherence therapeutic options might be divided into three groups based on their mechanism of action:
a) Molecules interfering with the physical properties of the red cell-endothelial adhesion process;
b) Molecules specifically interfering with sickle cell- endothelial adhesive mechanisms;
c) Molecules modulating inflammatory pathways involved in sickle cell endothelial adhesion;
d) Molecules affecting platelet function.
a) Molecules interfering with the physical properties of the red cell-endothelial adhesion process.
RheothRx (Poloxamer 188), a non-ionic surfactant copolymer was shown to
improve microvascular blood flow by lowering viscosity and frictional
forces. RheothRx was shown some beneficial effects on pain intensity
and duration of hospitalization in a pilot study on SCD patients
experiencing moderate to severe vaso- occlusive crisis.[96]
RheothRx was tested in a phase III clinical study for treatment of VOCs
in SCD. Although P188 has been shown to shorten the duration of pain
crisis, its effects on acute events were limited.[97,98]
Mast Therapeutics announced in 2016 negative results for a new phase
III trial with Vepoloxamer (MST-188), a IV agent tested to assess its
effect on reducing the duration of vaso-occlusive crises.[99]
b) Molecules interfering with sickle cell-endothelial adhesive mechanisms.
Recent studies in SCD have identified different mechanisms involved in
sickle cell-endothelium adhesive events, which may be of therapeutic
relevance (Figure 1): (i) the
integrin α4β1 receptor of fibronectin and the vascular adhesion
molecule-1 (VCAM-1), E-selectin and P-selectin; (ii) the thrombospondin
and/or collagen and receptor CD36, present on the surface of
endothelial cells, platelets and reticulocyte-rich subpopulations of
normal and sickle red cells; (iii) the sulfate glycolipids, which bind
thrombospondin, von-Willebrand factor multimer and laminin; (iv) the
Lutheran blood group proteins (BCAM/LU), whose expression is increased
in red cells from SCD patients; (v) the ICAM-4 (Landstein-Weiner blood
group glycoprotein-LW), which binds αVβ3 integrin receptors; and (vi)
the exposure of PS detectable in a subpopulation of sickle red cells,
which participates both in cell-cell adhesion to activated vascular
endothelium surface and in the activation of a coagulation system.
Monoclonal antibodies against the adhesion molecules or short synthetic
peptides interfering with ICAM-4 or αVβ3 integrin have been shown to
reduce adhesion events in SCD mouse models (Figure 1).
It is of interest to note that antibodies against adhesion molecules
block the heme induced vascular stasis, supporting again the connection
between heme, vasculopathy, and adhesion events in SCD.[68,100,101]
Among the agents interfering with red cell vascular adhesion events,
the blockade of adhesion mechanisms through interference with Selectins
seems to be a novel powerful therapeutic option for clinical management
of SCD. Selectins are a family of molecules mediating adhesion of blood
cells with activated vascular endothelial cells. and play a key role in
leukocyte recruitment as well as in sickle red cell adhesion to
inflammatory activated vascular endothelium. In addition, studies have
shown that P-selectin are increased in plasma of SCD patients.[65,102-106]
Different therapeutic strategies have been developed, to block
selectins: (i) pan-Selectin antagonist (GMI-1070, rivipansel); (ii)
humanized anti-P-Selectin antibody (SelG1, crinalizumab); (iii)
P-selectin-aptamer; and (iv) sevuparin.[11,12,15,50,65,104,107-112] Rivipansel
is a glycomimetic pan-selectin antagonist, which was tested in phase-I
and -II studies in SCD. Rivipansel showed a safe profile, reducing the
levels of E-Selectin in SCD patients during acute VOCs.[107,113]
In phase II study, rivipansel beneficially affected the number of pain
crisis in a small number of SCD subjects (#NCT01119833). However, these
data were obtained including some SC patients, which generates some
difficulties in their interpretation. An on-going phase II study is
focused on SCD children.
Crinalizumab is a humanized P-Selectin
antibody, which has been tested in a multinational double-blind
placebo-controlled trial (SUSTAIN, #NCT0185361).[15,111] SCD subjects (SS, SC, Sβ+ and Sβ0
genotype) were treated with crinalizumab either 2.5 or 5 mg/Kg every 4
weeks. Crinalizumab at the dosage of 5 mg/Kg every 4 weeks reduced the
number of pain crisis and increased the time between VOCs in SCD
independently from possible preceding HU treatment.[15,111,112]
An
additional strategy targeting P-Selectins is represented by the use of
low molecular weight heparins, such as tinzaparin, which has been shown
to block the P-Selectin system and to reduce the duration and the
severity of VOCs in few cases of SCD patients.[12,50] Sevuparin is a derivative of low-molecular weight heparin, lacking anticoagulant activity and it has been evaluated in SCD.[109,114]
Sevuparin acts on multiple targets: (i) P and L-selectins; (ii)
thrombospondin- Fibronectin-Von Willebrand factor; and (iii) sickle-
leukocyte-endothelial cells interaction. A phase II multicenter
international trial on sevuparin in acute VOCs is ongoing.
c) Molecules modulating inflammatory pathways involved in sickle cell endothelial adhesion.
Another set of novel therapeutic option is represented by agents
modulating the inflammatory pathways that participate to adhesion
events in SCD.
Studies in different models of
hypoxia/reoxygenation stress have shown that adenosine is released from
cells and interacts with A (1-3) receptors (AR), which are present on
endothelial cells, leukocytes and iNKT cells. This promotes the
activation of the transcriptional factor NF-kB, which orchestrates the
inflammatory response. iNKT are a subgroup of T lymphocytes that
affects both innate and adaptive immunity, participating to
inflammatory cascade.[115-117]
ncreased iNKT circulating cells have been observed in
SCD subjects on both steady state and during acute VOCs. Antibodies
against iNKT cells (NKTT120) have been developed, based on the key role
that adhesion and inflammation are involved in the pathogenesis of
severe acute complication of SCD (#NCT01783691).[15,99,115]
Field et al. recently reported the failure of regadenoson in reducing
iNKT activation and in interfering with the severity of the acute
clinical manifestations of SCD patients enrolled in randomized phase II
clinical trial (#NCT01788631).[118]
An attempt
to target inflammatory vasculopathy and to modulate inflammatory
response has been made based on the evidences in other diseases such as
in cardiovascular disease looking to dietary manipulation with omega-3
fatty acids (ω-3 PUFAs). Supplementation with omega-3 fatty acids has
been reported to (i) beneficially affect red cell membrane lipid
composition; (ii) modulate soluble and cellular inflammatory response
and coagulation cascade; and (iii) to favor NO production.[119-122]
In SCD, the fatty acid profile of sickle erythrocytes is altered
compared to healthy controls, with a relative increase in the ratio of
ω-6 to ω-3 PUFAs, in agreement with sustained chronic inflammation.[123,124]
In humanized mouse model for SCD, PUFA supplementation protects against
acute sickle cell-related lung and liver damages during
hypoxia/reoxygenation induced VOCs.[14] A phase II
multicenter randomized double-blind placebo- controlled study in SCD
patients reported that ω-3 fatty acid supplementation reduced pain
episode in SCD subjects (SCOT, #NCT02973360).[125-128]
d) Molecules affecting platelet function.
The role of platelets in clinical manifestations of SCD on both steady
state and acute events has been only partially characterized and much
still remains to be investigated.[5,11,50]
Early evidence on the beneficial effects of ticlopidine on reducing the
rate of pain crisis highlighted the potential role of platelet
activation and aggregation during acute events in SCD.[129]
However, a multicentric phase 2 study on prasugrel, a third- generation
anti-platelet agent, in adult with SCD showed a reduction of platelet
activation without change in pain rate.[130]
Recently, ticagrelor, a direct anti-platelet agent with some effects on
vascular tone and inflammatory response has been evaluated in a
dose-finding study on SCD children (HESTIA1, #NCT02214121).[11,131]
Ticagrelor was well tolerated without significant drug related adverse
events, in particular no hemorrhagic events were reported. Noteworthy,
in SCD children ticagrelor induced platelet inhibition similar to that
reported in adults with acute coronary disease.[131] A phase III clinical trial with ticagrelor in adults with SCD is on-going (#NCT02482298).[11,131]
Anti-Oxidant Agents and Sickle Cell Disease.
SCD is also characterized by a highly pro-oxidant environment due to
the elevated production of reactive oxygen species (ROS) generated by
increased levels of pathological free heme and iron and a reduction in
anti- oxidant systems such as GSH (Figure1).[5,7,12,132,133]
N- Acetyl-Cysteine (NAC), an exogenous thiol donor, has been studied
both in vitro and in vivo in SCD patients. NAC supplementation
(1,200-2,400 mg/day) was shown to reduce the formation of dense red
cells and the rate of hemolysis and to increase GSH levels in SCD
subjects. However, Sins et al. have recently reported a randomized,
placebo-, double-blind trial (#NCT01849016) on NAC in SCD. Although the
study shows a failure of NAC in affecting acute clinical manifestations
of SCD, the Authors point out that the low adherence of SCD to NAC
treatment might be responsible for the reduced biological effect of NAC
in SCD. A clinical trial with high dose of NAC during acute VOCs
related to SCD is ongoing (#NCT 01800526).[134-136]
L-Glutamine
is a likely anti-oxidant agent in SCD. Glutamine is involved in GSH
metabolism since it preserves NADPH levels required for GSH recycling,
and it is the precursor for nicotinamide adenine dinucleotide (NAD) and
arginine.[137-139] A first randomized, double blind,
placebo-controlled parallel group trial with L-glutamine
supplementation in SCD patients showed reduction in number of
hospitalization compared to historic patients data.[138]
Recently, a multicenter, randomize, placebo-controlled double- blind
phase III clinical trial with L-glutamine (0.3 g/Kg twice a day)
involving 230 SS/Sbeta0 patients with > 2 pain crisis showed that
L-glutamine supplementation reduced the mean number and length of
hospitalization, associated with increased median time to the first
crisis.[137] Both studies have several
limitations such as (i) the high rate of patient drop-out; (ii) the
presence of fatal events due to multiorgan failure in L-glutamine arm;
(iii) the lack of effects on hematologic parameters and hemolytic
indices; and (iv) the absence of clear data on L-glutamine mechanism of
action.[137,140] Since no
information are available on log-term use of L- glutamine
supplementation as well on the systemic effects of L-glutamine, the
sickle cell scientific community should use caution in prescribing L-
glutamine supplement for both adult and pediatric SCD patients.[140]
Future studies are required to further define the role of anti-oxidant
treatments in the clinical management of SCD subjects.
Curative Options in Sickle Cell Disease
In
the last two decades, progresses on hematopoietic stem cell
transplantation (HSCT) strategies have allowed to offer a new curative
option to patients with SCD. The major limitation in diffusion of HSCT
is (i) the availability of leukocyte antigen (HLA)-matched sibling
donor; (ii) the toxicities associated with myeloablative conditioning;
and (iii) inflammatory vasculopathy.[141-145]
Recently, lentiviral gene therapy has been reported to be safe and to
positively impact hematologic phenotype in a child with SCD.[146] Different clinical trials on gene therapy in SCD are on-going in various countries.[141-144]
Finally,
the development of CRISPR/Cas9 genome editing (GE) strategy has been
reported to represent a new potential therapeutic tool for genetic
correction of SCD.[147-149] However, in SCD GE is still limited to cell- and/or animal-based studies.
Conclusions
In
conclusion, the emerging picture for new treatment of SCD is that
formation of dense red cells, vasculopathy, adhesion events and
inflammation as well as oxidative stress might constitute new
pharmacological targets (Figure 3).
Promising
data have been reported on new therapeutic tools interfering with
P-selectin and modulating inflammatory vasculopathy. However, some
concerns have been expressed about possible reductions of appropriate
inflammatory responses to pathogens, although the initial trials did
not show any signal in this direction. A new field of combinatorial
therapy for SCD will require a holistic approach, considering the
improvement of patient quality of life as an important outcome in
designing new clinical studies.
Acknowledgments
We
would like to thank Dr Carlo Brugnara (Boston Children’s Hospital,
Harvard Medical School, Boston, MA; USA) for fruitful discussion and
manuscript revision.
Competing Interests and Funding
The Authors declare that they have no conflict of interest. This work was supported by FUR-UNIVR (LDF).
References
- Modell B, Darlison M. Global epidemiology of
haemoglobin disorders and derived service indicators. Bulletin of the
World Health Organization 2008;86:480-487. https://doi.org/10.2471/BLT.06.036673 PMid:18568278 PMCid:PMC2647473
- Murray
CJ, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for
291 diseases and injuries in 21 regions, 1990-2010: a systematic
analysis for the Global Burden of Disease Study 2010. Lancet
2012;380:2197-2223. https://doi.org/10.1016/S0140-6736(12)61689-4
- Weatherall
DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global
health problem. Bulletin of the World Health Organization
2001;79:704-712. PMid:11545326 PMCid:PMC2566499
- Piel
FB, Patil AP, Howes RE, et al. Global epidemiology of sickle
haemoglobin in neonates: a contemporary geostatistical model-based map
and population estimates. Lancet 2013;381:142-151. https://doi.org/10.1016/S0140-6736(12)61229-X
- De Franceschi L, Cappellini MD, Olivieri O. Thrombosis and sickle cell disease. Semin Thromb Hemost 2011;37:226-236. https://doi.org/10.1055/s-0031-1273087 PMid:21455857
- Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Advances in protein chemistry 1990;40:63-279. https://doi.org/10.1016/S0065-3233(08)60287-9
- De
Franceschi L, Corrocher R. Established and experimental treatments for
sickle cell disease. Haematologica 2004;89:348-356. PMid:15020275
- Ballas
SK, Smith ED. Red blood cell changes during the evolution of the sickle
cell painful crisis. Blood 1992;79:2154-2163. PMid:1562742
- Vinchi
F, De Franceschi L, Ghigo A, et al. Hemopexin therapy improves
cardiovascular function by preventing heme-induced endothelial toxicity
in mouse models of hemolytic diseases. Circulation 2013;127:1317-1329. https://doi.org/10.1161/CIRCULATIONAHA.112.130179 PMid:23446829
- Hebbel
RP, Vercellotti G, Nath KA. A systems biology consideration of the
vasculopathy of sickle cell anemia: the need for multi-modality
chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets
2009;9:271-292. https://doi.org/10.2174/1871529X10909040271 PMid:19751187 PMCid:PMC2914570
- Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood 2016;127:810-819. https://doi.org/10.1182/blood-2015-09-618553 PMid:26758919 PMCid:PMC4760087
- Manwani
D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology
and novel targeted therapies. Blood 2013;122:3892-3898. https://doi.org/10.1182/blood-2013-05-498311 PMid:24052549 PMCid:PMC3854110
- Hebbel RP. Adhesion of sickle red cells to endothelium: myths and future directions. Transfus Clin Biol 2008;15:14-18. https://doi.org/10.1016/j.tracli.2008.03.011 PMid:18501652
- Kalish
BT, Matte A, Andolfo I, et al. Dietary omega-3 fatty acids protect
against vasculopathy in a transgenic mouse model of sickle cell
disease. Haematologica 2015;100:870-880. https://doi.org/10.3324/haematol.2015.124586 PMid:25934765 PMCid:PMC4486221
- Ataga
KI, Kutlar A, Kanter J, et al. Crizanlizumab for the Prevention of Pain
Crises in Sickle Cell Disease. N Engl J Med 2017;376:429-439. https://doi.org/10.1056/NEJMoa1611770 PMid:27959701 PMCid:PMC5481200
- Hidalgo
A, Chang J, Jang JE, et al. Heterotypic interactions enabled by
polarized neutrophil microdomains mediate thromboinflammatory injury.
Nature medicine 2009;15:384-391. https://doi.org/10.1038/nm.1939 PMid:19305412 PMCid:PMC2772164
- Dalle
Carbonare L, Matte A, Valenti MT, et al. Hypoxia-reperfusion affects
osteogenic lineage and promotes sickle cell bone disease. Blood
2015;126:2320-2328. https://doi.org/10.1182/blood-2015-04-641969 PMid:26330244
- Sabaa
N, de Franceschi L, Bonnin P, et al. Endothelin receptor antagonism
prevents hypoxia-induced mortality and morbidity in a mouse model of
sickle-cell disease. J Clin Invest 2008;118:1924-1933. https://doi.org/10.1172/JCI33308 PMid:18382768 PMCid:PMC2276396
- Wieschhaus
A, Khan A, Zaidi A, et al. Calpain-1 knockout reveals broad effects on
erythrocyte deformability and physiology. Biochem J 2012;448:141-152. https://doi.org/10.1042/BJ20121008 PMid:22870887 PMCid:PMC3955119
- Siciliano
A, Turrini F, Bertoldi M, et al. Deoxygenation affects tyrosine
phosphoproteome of red cell membrane from patients with sickle cell
disease. Blood Cells Mol Dis 2010;44:233-242. https://doi.org/10.1016/j.bcmd.2010.02.007 PMid:20206558
- Yawn
BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell
disease: summary of the 2014 evidence-based report by expert panel
members. JAMA 2014;312:1033-1048. https://doi.org/10.1001/jama.2014.10517 PMid:25203083
- Engert
A, Balduini C, Brand A, et al. The European Hematology Association
Roadmap for European Hematology Research: a consensus document.
Haematologica 2016;101:115-208. https://doi.org/10.3324/haematol.2015.136739 PMid:26819058 PMCid:PMC4938336
- Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med 2008;358:1362-1369. https://doi.org/10.1056/NEJMct0708272 PMid:18367739
- Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol 1992;19:1-10. PMid:1641648
- Charache
S. Mechanism of action of hydroxyurea in the management of sickle cell
anemia in adults. Semin Hematol 1997;34:15-21. PMid:9317197
- Charache
S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency
of painful crises in sickle cell anemia. Investigators of the
Multicenter Study of Hydroxyurea in Sickle Cell Anemia [see comments].
New England Journal of Medicine 1995;332:1317-1322. https://doi.org/10.1056/NEJM199505183322001 PMid:7715639
- Saleh
AW, Hillen HF, Duits AJ. Levels of endothelial, neutrophil and
platelet-specific factors in sickle cell anemia patients during
hydroxyurea therapy. Acta Haematol 1999;102:31-37. https://doi.org/10.1159/000040964 PMid:10473885
- Ware RE, de Montalembert M, Tshilolo L, et al. Sickle cell disease. Lancet 2017;390:311-323. https://doi.org/10.1016/S0140-6736(17)30193-9
- Rigano
P, De Franceschi L, Sainati L, et al. Real-life experience with
hydroxyurea in sickle cell disease: A multicenter study in a cohort of
patients with heterogeneous descent. Blood Cells Mol Dis 2018;69:82-89.
https://doi.org/10.1016/j.bcmd.2017.08.017 PMid:29107441
- Pule
GD, Mowla S, Novitzky N, et al. A systematic review of known mechanisms
of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell
disease. Expert Rev Hematol 2015;8:669-679. https://doi.org/10.1586/17474086.2015.1078235 PMid:26327494 PMCid:PMC4829639
- Jison
ML, Munson PJ, Barb JJ, et al. Blood mononuclear cell gene expression
profiles characterize the oxidant, hemolytic, and inflammatory stress
of sickle cell disease. Blood 2004;104:270-280. https://doi.org/10.1182/blood-2003-08-2760 PMid:15031206 PMCid:PMC5560446
- Stettler
N, McKiernan CM, Melin CQ, et al. Proportion of adults with sickle cell
anemia and pain crises receiving hydroxyurea. JAMA 2015;313:1671-1672. https://doi.org/10.1001/jama.2015.3075 PMid:25919532
- Wong TE, Brandow AM, Lim W, et al. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014;124:3850-3857. https://doi.org/10.1182/blood-2014-08-435768 PMid:25287707 PMCid:PMC4271176
- Crosby
WH, Dameshek W. The significance of hemoglobinemia and associated
hemosideriinuria, with particular references to various types of
hemolytic anemia. J Lab Clin Med 1951;38:829. PMid:14889070
- Voskaridou
E, Christoulas D, Bilalis A, et al. The effect of prolonged
administration of hydroxyurea on morbidity and mortality in adult
patients with sickle cell syndromes: results of a 17-year,
single-center trial (LaSHS). Blood 2010;115:2354-2363. https://doi.org/10.1182/blood-2009-05-221333 PMid:19903897
- Wang
WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children
with sickle-cell anaemia: a multicentre, randomised, controlled trial
(BABY HUG). Lancet 2011;377:1663-1672. https://doi.org/10.1016/S0140-6736(11)60355-3
- Brousse
V, Gandhi S, de Montalembert M, et al. Combined blood transfusion and
hydroxycarbamide in children with sickle cell anaemia. Br J Haematol
2013;160:259-261. https://doi.org/10.1111/bjh.12104 PMid:23116405
- Bernaudin
F, Verlhac S, Arnaud C, et al. Long-term treatment follow-up of
children with sickle cell disease monitored with abnormal transcranial
Doppler velocities. Blood 2016;127:1814-1822. https://doi.org/10.1182/blood-2015-10-675231 PMid:26851292
- Ware
RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic
transfusion for maintenance of transcranial doppler flow velocities in
children with sickle cell anaemia-TCD With Transfusions Changing to
Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3,
non-inferiority trial. Lancet 2016;387:661-670. https://doi.org/10.1016/S0140-6736(15)01041-7
- Helton
KJ, Adams RJ, Kesler KL, et al. Magnetic resonance imaging/angiography
and transcranial Doppler velocities in sickle cell anemia: results from
the SWiTCH trial. Blood 2014;124:891-898.
https://doi.org/10.1182/blood-2013-12-545186 PMid:24914136 PMCid:PMC4126329
- Inoue
S, Kodjebacheva G, Scherrer T, et al. Adherence to hydroxyurea
medication by children with sickle cell disease (SCD) using an
electronic device: a feasibility study. Int J Hematol 2016;104:200-207.
https://doi.org/10.1007/s12185-016-2027-x PMid:27225236
- Han
J, Bhat S, Gowhari M, et al. Impact of a Clinical Pharmacy Service on
the Management of Patients in a Sickle Cell Disease Outpatient Center.
Pharmacotherapy 2016;36:1166-1172. https://doi.org/10.1002/phar.1834 PMid:27639254 PMCid:PMC5373798
- Green
NS, Manwani D, Qureshi M, et al. Decreased fetal hemoglobin over time
among youth with sickle cell disease on hydroxyurea is associated with
higher urgent hospital use. Pediatr Blood Cancer 2016;63:2146-2153. https://doi.org/10.1002/pbc.26161 PMid:27573582 PMCid:PMC5072999
- Smaldone
A, Findley S, Manwani D, et al. HABIT, a Randomized Feasibility Trial
to Increase Hydroxyurea Adherence, Suggests Improved Health-Related
Quality of Life in Youths with Sickle Cell Disease. J Pediatr
2018;197:177-185 e172.
- Ansong D, Akoto
AO, Ocloo D, et al. Sickle cell disease: management options and
challenges in developing countries. Mediterr J Hematol Infect Dis
2013;5:e2013062. https://doi.org/10.4084/mjhid.2013.062 PMid:24363877 PMCid:PMC3867228
- Opoka
RO, Ndugwa CM, Latham TS, et al. Novel use Of Hydroxyurea in an African
Region with Malaria (NOHARM): a trial for children with sickle cell
anemia. Blood 2017;130:2585-2593. https://doi.org/10.1182/blood-2017-06-788935 PMid:29051184
- Tayo
BO, Akingbola TS, Saraf SL, et al. Fixed Low-Dose Hydroxyurea for the
Treatment of Adults with Sickle Cell Anemia in Nigeria. Am J Hematol
2018.
- De Franceschi L. Pathophisiology
of sickle cell disease and new drugs for the treatment. Mediterr J
Hematol Infect Dis 2009;1:e2009024.
- Stocker
JW, De Franceschi L, McNaughton-Smith GA, et al. ICA-17043, a novel
Gardos channel blocker, prevents sickled red blood cell dehydration in
vitro and in vivo in SAD mice. Blood 2003;101:2412-2418. https://doi.org/10.1182/blood-2002-05-1433 PMid:12433690
- De
Franceschi L, Saadane N, Trudel M, et al. Treatment with oral
clotrimazole blocks Ca(2+)-activated K+ transport and reverses
erythrocyte dehydration in transgenic SAD mice. A model for therapy of
sickle cell disease. J Clin Invest 1994;93:1670-1676. https://doi.org/10.1172/JCI117149 PMid:7512989 PMCid:PMC294212
- Telen MJ. Developing new pharmacotherapeutic approaches to treating sickle-cell disease. ISBT Sci Ser 2017;12:239-247. https://doi.org/10.1111/voxs.12305 PMid:28484512 PMCid:PMC5418585
- De
Franceschi L, Franco RS, Bertoldi M, et al. Pharmacological inhibition
of calpain-1 prevents red cell dehydration and reduces Gardos channel
activity in a mouse model of sickle cell disease. FASEB J
2013;27:750-759. https://doi.org/10.1096/fj.12-217836 PMid:23085996 PMCid:PMC3545531
- McNaughton-Smith
GA, Burns JF, Stocker JW, et al. Novel inhibitors of the Gardos channel
for the treatment of sickle cell disease. J Med Chem 2008;51:976-982. https://doi.org/10.1021/jm070663s PMid:18232633
- De
Franceschi L, Brugnara C, Rouyer-Fessard P, et al. Formation of dense
erythrocytes in SAD mice exposed to chronic hypoxia: evaluation of
different therapeutic regimens and of a combination of oral
clotrimazole and magnesium therapies. Blood 1999;94:4307-4313.
PMid:10590075
- Li Q, Henry ER,
Hofrichter J, et al. Kinetic assay shows that increasing red cell
volume could be a treatment for sickle cell disease. Proc Natl Acad Sci
U S A 2017;114:E689-E696. https://doi.org/10.1073/pnas.1619054114 PMid:28096387 PMCid:PMC5293101
- Dufu
K, Oksenberg D. GBT440 reverses sickling of sickled red blood cells
under hypoxic conditions in vitro. Hematol Rep 2018;10:7419. https://doi.org/10.4081/hr.2018.7419 PMid:30046411 PMCid:PMC6036981
- Metcalf
B, Chuang C, Dufu K, et al. Discovery of GBT440, an Orally Bioavailable
R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med Chem Lett
2017;8:321-326. https://doi.org/10.1021/acsmedchemlett.6b00491 PMid:28337324 PMCid:PMC5346980
- Oksenberg
D, Dufu K, Patel MP, et al. GBT440 increases haemoglobin oxygen
affinity, reduces sickling and prolongs RBC half-life in a murine model
of sickle cell disease. Br J Haematol 2016;175:141-153. https://doi.org/10.1111/bjh.14214 PMid:27378309
- Dufu
K OD, Zhou C, Hutchaleelaha A, Archer DR. GTx011, a potent allosteric
modifier of hemoglobin oxygen affinity, prevents RBC sickling in whole
blood and prolongs RNC half-life in vivo in a murine model of sickle
cell disease. In: Blood, editor. American Society of Hematology; 2014.
p a217.
- Patel M CP, Dufu
K, Metcalf B, Sinha U. GTx011, an anti-sickling compound, improves SS
blood rheology by reduction of HbS polymerization via allosteric
modulation of O2 affinity. In: Blood, editor. American Society of
Hematology 2014. p a1370.
- Blyden G,
Bridges KR, Bronte L. Case series of patients with severe sickle cell
disease treated with voxelotor (GBT440) by compassionate access. Am J
Hematol 2018.
- Telfer P, Agodoa I, Fox
KM, et al. Impact of voxelotor (GBT440) on unconjugated bilirubin and
jaundice in sickle cell disease. Hematol Rep 2018;10:7643. https://doi.org/10.4081/hr.2018.7643 PMid:30046415 PMCid:PMC6036983
- Estepp
JH. Voxelotor (GBT440), a first-in-class hemoglobin oxygen-affinity
modulator, has promising and reassuring preclinical and clinical data.
Am J Hematol 2018;93:326-329. https://doi.org/10.1002/ajh.25042 PMid:29352729
- Kato
GJ, Hebbel RP, Steinberg MH, et al. Vasculopathy in sickle cell
disease: Biology, pathophysiology, genetics, translational medicine,
and new research directions. Am J Hematol 2009;84:618-625. https://doi.org/10.1002/ajh.21475 PMid:19610078 PMCid:PMC3209715
- Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers 2018;4:18010. https://doi.org/10.1038/nrdp.2018.10 PMid:29542687
- Solovey
A, Gui L, Ramakrishnan S, et al. Sickle cell anemia as a possible state
of enhanced anti-apoptotic tone: survival effect of vascular
endothelial growth factor on circulating and unanchored endothelial
cells. Blood 1999;93:3824-3830. PMid:10339489
- Schaer
DJ, Buehler PW, Alayash AI, et al. Hemolysis and free hemoglobin
revisited: exploring hemoglobin and hemin scavengers as a novel class
of therapeutic proteins. Blood 2013;121:1276-1284. https://doi.org/10.1182/blood-2012-11-451229 PMid:23264591 PMCid:PMC3578950
- Belcher
JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to
endothelial cell activation and vaso-occlusion in murine sickle cell
disease. Blood 2014;123:377-390. https://doi.org/10.1182/blood-2013-04-495887 PMid:24277079 PMCid:PMC3894494
- Reiter
CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric
oxide bioavailability in sickle-cell disease. Nat Med 2002;8:1383-1389.
https://doi.org/10.1038/nm1202-799 PMid:12426562
- Belcher
JD, Vineyard JV, Bruzzone CM, et al. Heme oxygenase-1 gene delivery by
Sleeping Beauty inhibits vascular stasis in a murine model of sickle
cell disease. J Mol Med (Berl) 2010;88:665-675. https://doi.org/10.1007/s00109-010-0613-6 PMid:20306336 PMCid:PMC2877767
- Belcher JD, Beckman JD, Balla G, et al. Heme degradation and vascular injury. Antioxid Redox Signal 2010;12:233-248. https://doi.org/10.1089/ars.2009.2822 PMid:19697995 PMCid:PMC2821146
- Muller-Eberhard
U, Javid J, Liem HH, et al. Plasma concentrations of hemopexin,
haptoglobin and heme in patients with various hemolytic diseases. Blood
1968;32:811-815. PMid:5687939
- Ballas
SK, Marcolina MJ. Hyperhemolysis during the evolution of uncomplicated
acute painful episodes in patients with sickle cell anemia. Transfusion
2006;46:105-110. https://doi.org/10.1111/j.1537-2995.2006.00679.x PMid:16398738
- Ohga
S, Higashi E, Nomura A, et al. Haptoglobin therapy for acute favism: a
Japanese boy with glucose-6-phosphate dehydrogenase Guadalajara. Br J
Haematol 1995;89:421-423. https://doi.org/10.1111/j.1365-2141.1995.tb03322.x PMid:7873396
- Merle
NS, Grunenwald A, Rajaratnam H, et al. Intravascular hemolysis
activates complement via cell-free heme and heme-loaded microvesicles.
JCI Insight 2018;3.
- de Franceschi L,
Malpeli G, Scarpa A, et al. Protective effects of S-nitrosoalbumin on
lung injury induced by hypoxia-reoxygenation in mouse model of sickle
cell disease. Am J Physiol Lung Cell Mol Physiol 2006;291:L457-465. https://doi.org/10.1152/ajplung.00462.2005 PMid:16603592
- Louie
JE, Anderson CJ, Fayaz MFK, et al. Case series supporting heme
detoxification via therapeutic plasma exchange in acute multiorgan
failure syndrome resistant to red blood cell exchange in sickle cell
disease. Transfusion 2018;58:470-479. https://doi.org/10.1111/trf.14407 PMid:29193101
- Kato
GJ, Gladwin MT. Evolution of novel small-molecule therapeutics
targeting sickle cell vasculopathy. JAMA 2008;300:2638-2646. https://doi.org/10.1001/jama.2008.598 PMid:19066384 PMCid:PMC2756016
- Belcher
JD, Young M, Chen C, et al. MP4CO, a pegylated hemoglobin saturated
with carbon monoxide, is a modulator of HO-1, inflammation, and
vaso-occlusion in transgenic sickle mice. Blood 2013;122:2757-2764. https://doi.org/10.1182/blood-2013-02-486282 PMid:23908468 PMCid:PMC4067504
- de
Franceschi L, Baron A, Scarpa A, et al. Inhaled nitric oxide protects
transgenic SAD mice from sickle cell disease-specific lung injury
induced by hypoxia/reoxygenation. Blood 2003;102:1087-1096. https://doi.org/10.1182/blood-2002-07-2135 PMid:12689931
- De
Franceschi L, Platt OS, Malpeli G, et al. Protective effects of
phosphodiesterase-4 (PDE-4) inhibition in the early phase of pulmonary
arterial hypertension in transgenic sickle cell mice. FASEB J
2008;22:1849-1860. https://doi.org/10.1096/fj.07-098921 PMid:18245171
- Gladwin
MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute
treatment of sickle cell pain crisis: a randomized controlled trial.
JAMA 2011;305:893-902. https://doi.org/10.1001/jama.2011.235 PMid:21364138 PMCid:PMC3403835
- Kim-Shapiro
DB, Gladwin MT. Nitric oxide pathology and therapeutics in sickle cell
disease. Clin Hemorheol Microcirc 2018;68:223-237. https://doi.org/10.3233/CH-189009 PMid:29614634
- Bakshi
N, Morris CR. The role of the arginine metabolome in pain: implications
for sickle cell disease. Journal of pain research 2016;9:167-175.
PMid:27099528 PMCid:PMC4821376
- Morris
CR. Alterations of the arginine metabolome in sickle cell disease: a
growing rationale for arginine therapy. Hematology/ Oncology Clinics of
North America 2014;28:301-321. https://doi.org/10.1016/j.hoc.2013.11.008 PMid:24589268
- Weiner
DL, Hibberd PL, Betit P, et al. Preliminary assessment of inhaled
nitric oxide for acute vaso-occlusive crisis in pediatric patients with
sickle cell disease. JAMA 2003;289:1136-1142. https://doi.org/10.1001/jama.289.9.1136 PMid:12622584
- Head
CA, Swerdlow P, McDade WA, et al. Beneficial effects of nitric oxide
breathing in adult patients with sickle cell crisis. Am J Hematol
2010;85:800-802. https://doi.org/10.1002/ajh.21832 PMid:20799359
- Abid
S, Kebe K, Houssaini A, et al. New Nitric Oxide Donor NCX 1443:
Therapeutic Effects on Pulmonary Hypertension in the SAD Mouse Model of
Sickle Cell Disease. J Cardiovasc Pharmacol 2018;71:283-292. https://doi.org/10.1097/FJC.0000000000000570 PMid:29438213
- Morris
CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism,
hemolysis-associated pulmonary hypertension, and mortality in sickle
cell disease. JAMA 2005;294:81-90. https://doi.org/10.1001/jama.294.1.81 PMid:15998894 PMCid:PMC2065861
- Tharaux
PL, Hagege I, Placier S, et al. Urinary endothelin-1 as a marker of
renal damage in sickle cell disease. Nephrol Dial Transplant
2005;20:2408-2413. https://doi.org/10.1093/ndt/gfi111 PMid:16144850
- Hammerman
SI, Kourembanas S, Conca TJ, et al. Endothelin-1 production during the
acute chest syndrome in sickle cell disease. American journal of
respiratory and critical care medicine 1997;156:280-285. https://doi.org/10.1164/ajrccm.156.1.9611085 PMid:9230761
- Koehl
B, Nivoit P, El Nemer W, et al. The endothelin B receptor plays a
crucial role in the adhesion of neutrophils to the endothelium in
sickle cell disease. Haematologica 2017;102:1161-1172. https://doi.org/10.3324/haematol.2016.156869 PMid:28385784 PMCid:PMC5566019
- Taylor
C, Kasztan M, Tao B, et al. Combined hydroxyurea and ETA receptor
blockade reduces renal injury in the humanized sickle cell mouse. Acta
Physiol (Oxf) 2018:e13178. https://doi.org/10.1111/apha.13178 PMid:30144292
- Smith
TP, Haymond T, Smith SN, et al. Evidence for the endothelin system as
an emerging therapeutic target for the treatment of chronic pain.
Journal of Pain Research 2014;7:531-545. https://doi.org/10.2147/JPR.S65923 PMid:25210474 PMCid:PMC4155994
- Lutz
BM, Wu S, Gu X, et al. Endothelin type A receptors mediate pain in a
mouse model of sickle cell disease. Haematologica 2018;103:1124-1135. https://doi.org/10.3324/haematol.2017.187013 PMid:29545351 PMCid:PMC6029538
- Adams-Graves
P, Kedar A, Koshy M, et al. RheothRx (poloxamer 188) injection for the
acute painful episode of sickle cell disease: a pilot study. Blood
1997;90:2041-2046. PMid:9292541
- Orringer
EP, Casella JF, Ataga KI, et al. Purified poloxamer 188 for treatment
of acute vaso-occlusive crisis of sickle cell disease: A randomized
controlled trial. JAMA 2001;286:2099-2106. https://doi.org/10.1001/jama.286.17.2099 PMid:11694150
- Koo
S, Yang Y, Neu B. Poloxamer 188 reduces normal and
phosphatidylserine-exposing erythrocyte adhesion to endothelial cells
in dextran solutions. Colloids and surfaces B, Biointerfaces
2013;112:446-451. https://doi.org/10.1016/j.colsurfb.2013.07.035 PMid:24055859
- Archer N, Galacteros F, Brugnara C. 2015 Clinical trials update in sickle cell anemia. Am J Hematol 2015;90:934-950. https://doi.org/10.1002/ajh.24116 PMid:26178236 PMCid:PMC5752136
- Kaul
DK, Liu XD, Zhang X, et al. Peptides based on alphaV-binding domains of
erythrocyte ICAM-4 inhibit sickle red cell-endothelial interactions and
vaso-occlusion in the microcirculation. Am J Physiol Cell Physiol
2006;291:C922-930. https://doi.org/10.1152/ajpcell.00639.2005 PMid:16738001
- Kaul
DK, Liu XD, Zhang X, et al. Inhibition of sickle red cell adhesion and
vasoocclusion in the microcirculation by antioxidants. Am J Physiol
Heart Circ Physiol 2006;291:H167-175. https://doi.org/10.1152/ajpheart.01096.2005 PMid:16443674
- Pan
J, Xia L, McEver RP. Comparison of promoters for the murine and human
P-selectin genes suggests species-specific and conserved mechanisms for
transcriptional regulation in endothelial cells. J Biol Chem
1998;273:10058-10067. https://doi.org/10.1074/jbc.273.16.10058 PMid:9545353
- Matsui
NM, Borsig L, Rosen SD, et al. P-selectin mediates the adhesion of
sickle erythrocytes to the endothelium. Blood 2001;98:1955-1962. https://doi.org/10.1182/blood.V98.6.1955 PMid:11535535
- Kutlar
A, Ataga KI, McMahon L, et al. A potent oral P-selectin blocking agent
improves microcirculatory blood flow and a marker of endothelial cell
injury in patients with sickle cell disease. Am J Hematol
2012;87:536-539. https://doi.org/10.1002/ajh.23147 PMid:22488107
- Turhan
A, Weiss LA, Mohandas N, et al. Primary role for adherent leukocytes in
sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S
A 2002;99:3047-3051. https://doi.org/10.1073/pnas.052522799 PMid:11880644 PMCid:PMC122470
- Blann
AD, Mohan JS, Bareford D, et al. Soluble P-selectin and vascular
endothelial growth factor in steady state sickle cell disease:
relationship to genotype. J Thromb Thrombolysis 2008;25:185-189. https://doi.org/10.1007/s11239-007-0177-7 PMid:18080800
- Wun
T, Styles L, DeCastro L, et al. Phase 1 study of the E-selectin
inhibitor GMI 1070 in patients with sickle cell anemia. PLoS One
2014;9:e101301. https://doi.org/10.1371/journal.pone.0101301 PMid:24988449 PMCid:PMC4079300
- Chang
J, Patton JT, Sarkar A, et al. GMI-1070, a novel pan-selectin
antagonist, reverses acute vascular occlusions in sickle cell mice.
Blood 2010;116:1779-1786. https://doi.org/10.1182/blood-2009-12-260513 PMid:20508165 PMCid:PMC2947397
- Telen
MJ, Batchvarova M, Shan S, et al. Sevuparin binds to multiple adhesive
ligands and reduces sickle red blood cell-induced vaso-occlusion. Br J
Haematol 2016;175:935-948. https://doi.org/10.1111/bjh.14303 PMid:27549988
- Gutsaeva
DR, Parkerson JB, Yerigenahally SD, et al. Inhibition of cell adhesion
by anti-P-selectin aptamer: a new potential therapeutic agent for
sickle cell disease. Blood 2011;117:727-735. https://doi.org/10.1182/blood-2010-05-285718 PMid:20926770 PMCid:PMC3031491
- Ataga KI, Kutlar A, Kanter J. Crizanlizumab in Sickle Cell Disease. N Engl J Med 2017;376:1796. https://doi.org/10.1056/NEJMoa1611770 PMCid:PMC5481200
- Slomski A. Crizanlizumab Prevents Sickle Cell Pain Crises. JAMA 2017;317:798. https://doi.org/10.1001/jama.2017.0355
- Telen
MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in
SCD: reduction in time to resolution of vaso-occlusive events and
decreased opioid use. Blood 2015;125:2656-2664. https://doi.org/10.1182/blood-2014-06-583351 PMid:25733584 PMCid:PMC4408290
- White
J, Lindgren M, Liu K, et al. Sevuparin blocks sickle blood cell
adhesion and sickle-leucocyte rolling on immobilized L-selectin in a
dose dependent manner. Br J Haematol 2018.
- Field JJ, Nathan DG. Advances in sickle cell therapies in the hydroxyurea era. Mol Med 2014;20 Suppl 1:S37-42.
- Field
JJ, Ataga KI, Majerus E, Eaton CA, Mashal R, Nathan DG. A phase I
single ascending dose study of NKTT120 in stable adult sickle cell
patients. In: Blood, editor. American Society of Hematology; 2014. p
a977.
- Field JJ, Nathan DG, Linden J.
The role of adenosine signaling in sickle cell therapeutics.
Hematology/Oncology Clinics of North America 2014;28:287-299. https://doi.org/10.1016/j.hoc.2013.11.003 PMid:24589267 PMCid:PMC3997263
- Field
JJ, Majerus E, Gordeuk VR, et al. Randomized phase 2 trial of
regadenoson for treatment of acute vaso-occlusive crises in sickle cell
disease. Blood Adv 2017;1:1645-1649. https://doi.org/10.1182/bloodadvances.2017009613 PMid:29296811 PMCid:PMC5728341
- Massaro
M, Scoditti E, Carluccio MA, et al. Basic mechanisms behind the effects
of n-3 fatty acids on cardiovascular disease. Prostaglandins Leukot
Essent Fatty Acids 2008;79:109-115. https://doi.org/10.1016/j.plefa.2008.09.009 PMid:18951002
- Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients 2010;2:355-374. https://doi.org/10.3390/nu2030355 PMid:22254027 PMCid:PMC3257651
- Rangel-Huerta
OD, Aguilera CM, Mesa MD, et al. Omega-3 long-chain polyunsaturated
fatty acids supplementation on inflammatory biomakers: a systematic
review of randomised clinical trials. Br J Nutr 2012;107 Suppl
2:S159-170.
- Russo C, Olivieri O,
Girelli D, et al. Omega-3 polyunsaturated fatty acid supplements and
ambulatory blood pressure monitoring parameters in patients with mild
essential hypertension. J Hypertens 1995;13:1823-1826. https://doi.org/10.1097/00004872-199512010-00059 PMid:8903660
- Ren
H, Obike I, Okpala I, et al. Steady-state haemoglobin level in sickle
cell anaemia increases with an increase in erythrocyte membrane n-3
fatty acids. Prostaglandins Leukot Essent Fatty Acids 2005;72:415-421. https://doi.org/10.1016/j.plefa.2005.03.005 PMid:15876528
- Ren
H, Ghebremeskel K, Okpala I, et al. Abnormality of erythrocyte membrane
n-3 long chain polyunsaturated fatty acids in sickle cell haemoglobin C
(HbSC) disease is not as remarkable as in sickle cell anaemia (HbSS).
Prostaglandins Leukot Essent Fatty Acids 2006;74:1-6. https://doi.org/10.1016/j.plefa.2005.10.002 PMid:16314081
- Daak
AA, Ghebremeskel K, Hassan Z, et al. Effect of omega-3 (n-3) fatty acid
supplementation in patients with sickle cell anemia: randomized,
double-blind, placebo-controlled trial. Am J Clin Nutr 2013;97:37-44. https://doi.org/10.3945/ajcn.112.036319 PMid:23193009
- Daak
AA, Dampier CD, Fuh B, et al. Double-blind, randomized, multicenter
phase 2 study of SC411 in children with sickle cell disease (SCOT
trial). Blood Adv 2018;2:1969-1979. https://doi.org/10.1182/bloodadvances.2018021444 PMid:30097463 PMCid:PMC6093734
- Daak
A, Rabinowicz A, Ghebremeskel K. Omega-3 fatty acids are a potential
therapy for patients with sickle cell disease. Nat Rev Dis Primers
2018;4:15. https://doi.org/10.1038/s41572-018-0012-9 PMid:30093627
- Tomer
A, Kasey S, Connor WE, et al. Reduction of pain episodes and
prothrombotic activity in sickle cell disease by dietary n-3 fatty
acids. Thromb Haemost 2001;85:966-974. https://doi.org/10.1055/s-0037-1615948 PMid:11434703
- Cabannes
R, Lonsdorfer J, Castaigne JP, et al. Clinical and biological
double-blind-study of ticlopidine in preventive treatment of
sickle-cell disease crises. Agents and actions Supplements
1984;15:199-212. PMid:6385647
- Heeney
MM, Hoppe CC, Abboud MR, et al. A Multinational Trial of Prasugrel for
Sickle Cell Vaso-Occlusive Events. N Engl J Med 2016;374:625-635. https://doi.org/10.1056/NEJMoa1512021 PMid:26644172
- Hsu
LL, Sarnaik S, Williams S, et al. A dose-ranging study of ticagrelor in
children aged 3-17 years with sickle cell disease: a two-part phase 2
study. Am J Hematol 2018. https://doi.org/10.1002/ajh.25273
- Reid
M, Badaloo A, Forrester T, et al. In vivo rates of erythrocyte
glutathione synthesis in adults with sickle cell disease. American
journal of physiology Endocrinology and metabolism 2006;291:E73-79. https://doi.org/10.1152/ajpendo.00287.2005 PMid:16434557
- Silva
DG, Belini Junior E, de Almeida EA, et al. Oxidative stress in sickle
cell disease: an overview of erythrocyte redox metabolism and current
antioxidant therapeutic strategies. Free Radic Biol Med
2013;65:1101-1109. https://doi.org/10.1016/j.freeradbiomed.2013.08.181 PMid:24002011
- Joep
W.R. Sins XF, Karin Fijnvandraat, Melissa Dominguez, A. W. Rijneveld,
Jean-Louis Kerkhoffs, A van Meurs, M. R. De Groot, H Heijboer, Erfan
Nur, Brenda M Luken, Sacha S Zeerleder, Marie-Françoise Dresse,
Phu-Quoc Le, Philippe Hermans, Anna Vanderfaeillie, Eric Van Den Neste,
Fleur Samantha Benghiat, Rachel Kesse-Adu, Andre Delannoy, Andre Efira,
Marie-Agnes Azerad, C A de Borgie, Junmei Chen, Jose A. Lopez and Bart
J. Biemond. Effects of Oral N -Acetylcysteine on Oxidative Stress in
Patients with Sickle Cell Disease. In: Blood, editor. ASH. Atlanta;
2017.
- Sins JWR, Fijnvandraat K,
Rijneveld AW, et al. Effect of N-acetylcysteine on pain in daily life
in patients with sickle cell disease: a randomised clinical trial. Br J
Haematol 2017. PMid:28643376
- Joep
W.R. Sins KF, Anita W. Rijneveld, Martine B. Boom, Jean-Louis
Kerkhoffs, Alfred H. van Meurs, Marco R De Groot, Harriet Heijboer,
Marie-Françoise Dresse, Alina Ferster, Philippe Hermans, Anna
Vanderfaeillie, Eric W Van Den Neste, Fleur Samantha Benghiat, Jo
Howard, Rachel Kesse-Adu, Andre Delannoy, Andre Efira, Marie-Agnes
Azerad, Corianne A.J.M. de Borgie and Bart J. Biemond. N-Acetylcysteine
in Patients with Sickle Cell Disease: A Randomized Controlled Trial.
In: Blood, editor. ASH: Blood; 2016.
- Niihara Y, Miller ST, Kanter J, et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med 2018;379:226-235. https://doi.org/10.1056/NEJMoa1715971 PMid:30021096
- Niihara
Y KH, Tran L, Razon R, Macan H, Stark C, Wun T, Adams-Graves P. A phase
3 study of L-Glutamine Therapy for sickle cell anemia and sickle
b0-thalassemia. In: Blood, editor. American Society of Hematology:
Blood; 2014. p a86.
- Niihara Y, Zerez
CR, Akiyama DS, et al. Oral L-glutamine therapy for sickle cell anemia:
I. Subjective clinical improvement and favorable change in red cell NAD
redox potential. Am J Hematol 1998;58:117-121. https://doi.org/10.1002/(SICI)1096-8652(199806)58:2<117::AID-AJH5>3.0.CO;2-V
- Quinn CT. l-Glutamine for sickle cell anemia: more questions than answers. Blood 2018;132:689-693. https://doi.org/10.1182/blood-2018-03-834440
- Lagresle-Peyrou
C, Lefrere F, Magrin E, et al. Plerixafor enables safe, rapid,
efficient mobilization of hematopoietic stem cells in sickle cell
disease patients after exchange transfusion. Haematologica
2018;103:778-786. https://doi.org/10.3324/haematol.2017.184788 PMid:29472357 PMCid:PMC5927997
- Esrick EB, Bauer DE. Genetic therapies for sickle cell disease. Semin Hematol 2018;55:76-86. https://doi.org/10.1053/j.seminhematol.2018.04.014 PMid:29958563
- Leonard
A, Tisdale JF. Stem cell transplantation in sickle cell disease:
therapeutic potential and challenges faced. Expert Rev Hematol
2018;11:547-565. https://doi.org/10.1080/17474086.2018.1486703 PMid:29883237
- Angelucci
E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell
transplantation in thalassemia major and sickle cell disease:
indications and management recommendations from an international expert
panel. Haematologica 2014;99:811-820. https://doi.org/10.3324/haematol.2013.099747 PMid:24790059 PMCid:PMC4008115
- Saraf
SL, Oh AL, Patel PR, et al. Haploidentical Peripheral Blood Stem Cell
Transplantation Demonstrates Stable Engraftment in Adults with Sickle
Cell Disease. Biol Blood Marrow Transplant 2018;24:1759-1765. https://doi.org/10.1016/j.bbmt.2018.03.031 PMid:29656137
- Ribeil
JA, Hacein-Bey-Abina S, Payen E, et al. Gene Therapy in a Patient with
Sickle Cell Disease. N Engl J Med 2017;376:848-855. https://doi.org/10.1056/NEJMoa1609677 PMid:28249145
- Antoniani
C, Meneghini V, Lattanzi A, et al. Induction of fetal hemoglobin
synthesis by CRISPR/Cas9-mediated editing of the human beta-globin
locus. Blood 2018;131:1960-1973. https://doi.org/10.1182/blood-2017-10-811505 PMid:29519807
- Sato
M, Saitoh I, Inada E. Efficient CRISPR/Cas9-based gene correction in
induced pluripotent stem cells established from fibroblasts of patients
with sickle cell disease. Stem Cell Investig 2016;3:78. https://doi.org/10.21037/sci.2016.11.05 PMid:28066780 PMCid:PMC5182212
- Ye
L, Wang J, Tan Y, et al. Genome editing using CRISPR-Cas9 to create the
HPFH genotype in HSPCs: An approach for treating sickle cell disease
and beta-thalassemia. Proc Natl Acad Sci U S A 2016;113:10661-10665. https://doi.org/10.1073/pnas.1612075113 PMid:27601644 PMCid:PMC5035856
[TOP]