A. Al-Madhani1, A. Pathare2, S. Al Zadjali2, M. Al Rawahi2, I. Al-Nabhani2 and S. Alkindi2,3.
1 Department of Medicine, Sohar Hospital, Sohar, Oman.
2 Department of Haematology, Sultan Qaboos University Hospital, Oman.
3 College of Medicine & Health Sciences, Muscat, Oman.
Correspondence to: Dr. Salam Alkindi, BA, MB, BCh, BAO, MSc, FRCP,
Professor in Haematology and Consultant Haematologist. Department of
Haematology, College of Medicine & Health Sciences, Sultan Qaboos
University, P. O. Box 35, Muscat 123, Sultanate of Oman. Tel:
+96824141182, Fax: +96824144887. E-mail:
sskindi@yahoo.com
Published: January 1, 2019
Received: August 29, 2018
Accepted: November 5, 2018
Mediterr J Hematol Infect Dis 2019, 11(1): e2019005 DOI
10.4084/MJHID.2019.005
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Newborn cord blood screening identifies infants with underlying
haemoglobinopathies before they develop the characteristic symptoms or
sequelae.
Aims: This study was performed to validate the
interpretation high-performance chromatography (HPLC) along with
complete blood count (CBC) results as a tool for universal neonatal
screening of hemoglobin disorders in Oman.
Methods:
HPLC and CBC data on subjects who participated in the National Neonatal
screening program at birth were obtained from archival records. The
results recorded at birth were compared with a second study performed
on the same subjects, after approval from the local medical research
and ethics committee.
Results:
Only 290 subjects from amongst the original cohort of 3740 newborns
could be recalled between April 2010 to March 2011, to repeat HPLC and
CBC, as well as perform confirmatory DNA studies, wherever necessary.
All these subjects had been documented to show an initial abnormal
result. 31 cases who had no HbA at birth on HPLC were confirmed as
either homozygous β-thalassaemia major (n=5 subjects) or homozygous
sickle cell anemia (n=26 subjects) by appropriate DNA analysis.
Additionally, amongst 151 subjects, 72 subjects were studied in the
initial study by Hb Bart’s quantitation using the alpha thalassaemia
short program at birth. In this cohort, 42 subjects with Hb Bart’s
>1% at birth could be confirmed as having either deletional or
non-deletional thalassaemia by GAP PCR studies. No case of HbH was
detected in this cohort. Further, carrier status for structural
hemoglobin variants (HbS, HbC, HbD, HbE) (n=67) and beta thalassaemia
allele with low HbA at birth (n=29 out of 41) were confirmed by
relevant molecular studies.
Conclusions:
The study validated the earlier observation by 100% concordance with
the results of CBC and HPLC. Presence of Hb Bart’s at birth does not
always mean the presence of alpha thalassemia, as subjects with Hb
Bart’s below 1% by quantitation, were shown to be normal by molecular
studies.
|
Introduction
The
mortality rate in sickle cell disease (SCD) is highest in the first five
years of life and the greatest risk period is the second 6 month of
life, so early diagnosis of SCD by the newborn screening can have a
huge impact on the mortality and morbidity.[1-6] Early
detection of SCD in this period allows for the introduction of
penicillin, administration of recommended vaccinations along with
counseling & education of affected families, before the onset of
symptoms.
Oral penicillin prophylaxis in children with SCD
provided an impressive 85% reduction in the incidence of infections and
led to the reduction in morbidity and mortality of this disease in
childhood as there was a 30% fatality rate observed amongst children
with SCD who developed sepsis.[7,8] Vichinsky[9]
in 1988 had shown that the mortality rate was 1.8% for those diagnosed
in the newborn period, as compared to 8% amongst those patients
diagnosed after three months of age. Similarly, the Cooperative Study
of Sickle Cell Disease observed that by instituting prophylactic
penicillin, the incidence of sepsis was reduced to 8% and the mortality
rate to 25%.[1] Further evidence can be seen from the
Jamaican newborn cohort study, which showed a mortality rate of 25%
with only 30% of infants experiencing acute sequestration crisis.[10]
Health
education and genetic counseling are the two pillars of any genetic
screening program. A good example of a preventive program is when it is
coupled with health education as has been reported in a study by
Riddington C and Owusu-Ofori S.[7] This study showed
that 70% of parents were able to determine spleen size when proper
training was given to them, and it was also found that 21% of acute
sequestration crisis were diagnosed as a result of the mother's
examination. Furthermore, newborn screening programs in which a strong
parent education component was not incorporated have been unsuccessful
in reducing mortality. Thus, mortality rates of 30% and 14% have been
reported when no comprehensive medical follow- up was provided.[9]
The
early identification of haemoglobinopathies and initial referral of
those infants ensures prompt delivery of health care and allows
screening for markers of disease severity as well as the initiation of
prophylactic interventions before the development of clinical
complications. The parents can also be counselled about their plans for
the current and future children. Further, early detection of
thalassaemia major can allow for the monitoring of the development of
signs and symptoms of anemia and institute blood transfusions and
chelation promptly.
Different laboratory techniques are employed
for neonatal screening including isoelectric focusing (IEF), and HPLC.
IEF has the disadvantage of being a labor- intensive manual technique,
whereas the Bio-Rad’s Variant HPLC system (Bio-Rad Laboratories,
Hercules, CA, USA) is a rapid semi-automated system that is widely used
and is the backbone for screening hemoglobin variants at our
institution. There are several programs available for this system
including sickle cell short program, which is a rapid 3-min assay
capable of using either filter paper blood spots or whole blood
samples. This program is specifically designed to provide a qualitative
result for hemoglobins A, F, S, C, D, and E in the neonate. A second
program is the β-Thalassemia
Short Program, which is a 6.5-min assay designed to quantify HbA2 and
HbF, however confirmation of some hemoglobin variants at birth can be
difficult, in particular, carriers of β thalassemia until adequate HbA2
has developed.
Our study is aimed at validating the results
obtained at the initial testing of the neonates enrolled in the
national neonatal screening program[11] with a
simple, cost-effective HPLC and CBC. We were able to follow up 290 such
subjects who were initially tested in the national neonatal screening
program at birth and then recalled now for repeat testing, and use the
opportunity to validate the initial results of HPLC and CBC by
confirmatory molecular studies to document and ascertain the final
diagnosis.
Methods
This
prospective follow-up study was conducted at Sultan Qaboos University
Hospital throughout one year between April 2010 to March 2011. In this
study, 290 babies from the original cohort of 3740 newborns were
studied.[11] All children had an abnormal cord blood
screening test. The study was approved by the local Medical Research
& Ethics committee of the hospital.
5 ml of venous blood was
collected in vacutainer tube with K2EDTA anticoagulant. HPLC and CBC
were performed using this blood sample, and 2 ml blood was used for
obtaining genomic DNA according to the manufacturer’s instructions
using the QIAamp DNA Blood mini kit (Qiagen, Inc., Valencia, CA, USA).
HPLC
was performed within 12-24 hours of collection of a blood sample using
the Bio-Rad VARIANT™ instrument (Bio-Rad Laboratories, Hercules, CA,
USA) and the “β-thalassemia short program. A CBC was performed on Cell
Dyn 4000 automated blood cell counter (Abbott Diagnostics, Santa Clara,
CA, USA).
Hb Bart’s quantitation was performed using α-thalassaemia short program (Biorad Variant II) at birth (within 12-24 hours of collection).[11]
In this follow-up study, Hb Bart’s positive cases were initially
screened by Genescan technique to determine the next approach.[12] Multiplex Gap PCR procedure was used to detect the seven common deletions occurring in the α-globin gene cluster.[12]
Additionally, in subjects with no deletional defect (n=1), automated
direct nucleotide sequencing (ABI 3130; Applied Biosystems, Foster
City, CA, USA) of the selectively amplified alpha1 and alpha2 globin
genes was performed to characterize non- deletional α-thalassaemia determinants using appropriate primers to ascertain the nature of the underlying molecular defect.[13]
Statistical Analysis.
The data were archived on a Microsoft Excel Database on a dedicated
computer. All analysis was carried out by using the SPSS software (IBM
SPSS Inc., USA. Ver. 23). Normally distributed results were expressed
as mean value + SD, whereas, the non–parametric data as median
(interquartile range). Students’ t-test was used to compare the
statistical significance between the means of various groups. A p-
value <0.05 was considered as significant. HPLC sensitivity of Hb
Bart’s quantitation at birth was reported using the manufacturer's
values (alpha thalassaemia short program, Variant II) and tested by
appropriate molecular techniques.
Results
Only
290 subjects from amongst the original cohort of 3740 newborns could be
recalled between April 2010 to March 2011, to repeat HPLC and CBC, as
well as perform confirmatory DNA studies. This cohort could be
categorized into three groups according to the cord blood results at
birth.[11] Group A: Subjects with no haemoglobin A by
HPLC at birth (n=31); Group B: Subjects with Hb Bart’s at birth on HPLC
using the “β- thalassemia short program (n=151); Group C: Subjects with
abnormal qualitative or quantitative beta chain variants based on HPLC
at birth (n=108).
Group A comprised of 31 children with no adult
haemoglobin detected at birth by HPLC. These babies were all re-tested
between 3 to 6 months of age according to the current guidelines.
Repeat HPLC was consistent with homozygous β-thalassaemia major in 5
babies with a significantly high HbF and HbA2, whereas the remaining 26
babies HPLC showed an HbS values between 92-94% consistent with sickle
cell anaemia. Repeat red cell indices in comparison to cord blood
results (Table 1a) showed a
significant drop in Hemoglobin (p < 0.001), MCV (p < 0.0001) and
MCH (p < 0.001) with increase in RBC count (p < 0.01). In this
group, the earlier diagnosis by HPLC was re- confirmed in all 31 cases
by appropriate DNA analysis to document the underlying mutation.
Sequence analysis of the entire β-globin gene and the β-globin gene promoter region was amplified by polymerase chain reaction (PCR) using appropriate primers.[12]
In
Group B, there were 151 babies who were suspected to have α
Thalassaemia at birth based on the presence of Hb Bart’s on HPLC.[11]
Repeat red cell indices in this follow-up study showed that the
hemoglobin, RBC counts, and MCHC were with-in the age-related normal
ranges. However as expected, in comparison to cord blood results, there
was a significant drop in hemoglobin (p < 0.001), MCV (p < 0.001)
and MCH (p < 0.001) with increase in RBC count (p < 0.05).
There were no significant differences in MCHC (Table 1b).
On repeat HPLC, no abnormal haemoglobin variant was detected. Further,
the fetal hemoglobin and adult haemoglobin were different but
appropriate for age. Table 1c & d
show comparison of red cell indices at birth and at follow-up study in
group B cohort subjects with one alpha gene deletion (n=20) and two
alpha gene deletions (n=22) with similar observations as above
respectively.
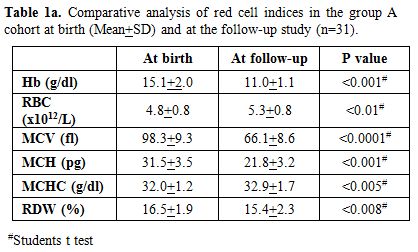 |
Table 1a. Comparative analysis of red cell indices in the group A cohort at birth (Mean+SD) and at the follow-up study (n=31). |
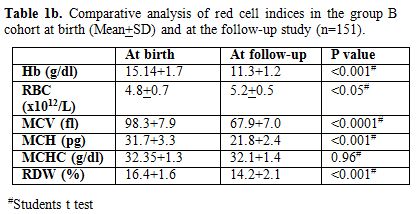 |
Table 1b. Comparative analysis of red cell indices in the group B cohort at birth (Mean+SD) and at the follow-up study (n=151). |
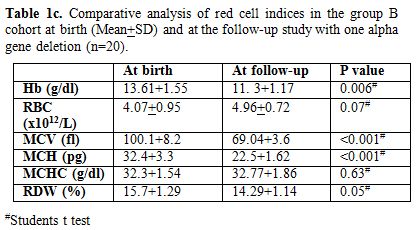 |
Table 1c. Comparative
analysis of red cell indices in the group B cohort at birth (Mean+SD)
and at the follow-up study with one alpha gene deletion (n=20). |
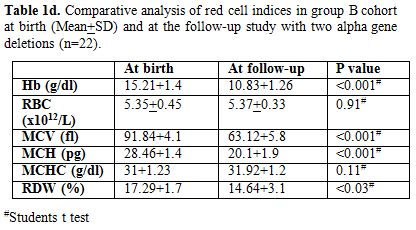 |
Table1d. Comparative analysis of red cell
indices in group B cohort at birth (Mean+SD) and at the follow-up study
with two alpha gene deletions (n=22). |
Amongst
these 151 subjects who had Hb Bart’s at birth, only 72 subjects had
been earlier studied by Hb Bart’s quantitation using α
alpha thalassaemia short program (Biorad Variant II) at birth. Hb
Bart’s was (< 1%) in 30 babies; between >1 to <3% in 20
babies, and >3% in twenty-two babies (Table 2a and 2b) (Figures 1 and 2). Table 2a
shows the comparative analysis of the various red cell indices at birth
and follow-up. Most of the parameters were significantly altered except
MCHC. MCV followed by MCH were the most important discriminators
reflecting the microcytic hypochromic red cell maturation. Multiplex
GAP PCR in the recalled subjects correctly identified the presence of
alpha thalassaemia in subjects that had more than 1% Hb Bart’s at birth
(n=42).
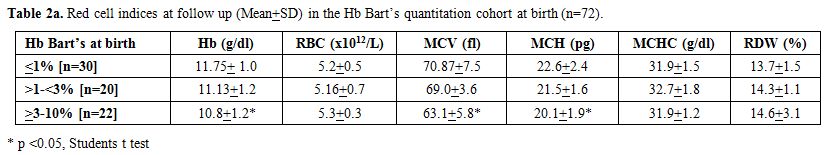 |
Table 2a.
Red cell indices at follow up (Mean+SD) in the Hb Bart’s quantitation cohort at birth (n=72). |
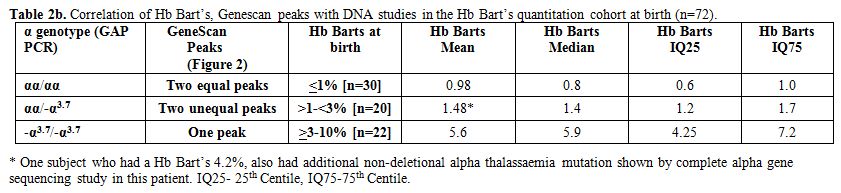 |
Table 2b. Correlation of Hb Bart’s, Genescan peaks with DNA studies in the Hb Bart’s quantitation cohort at birth (n=72). |
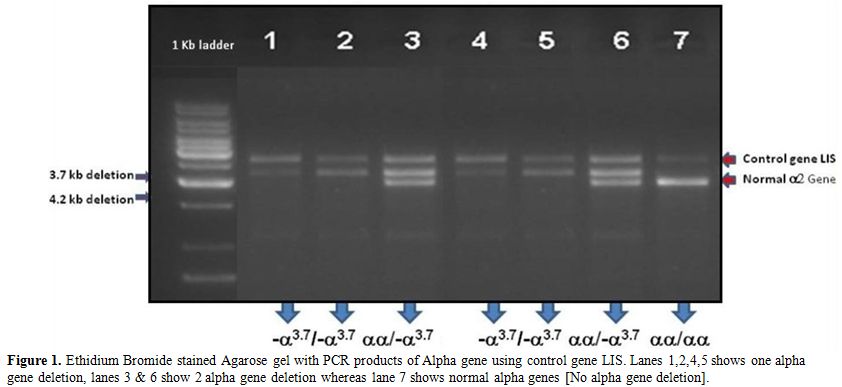 |
Figure 1. Ethidium Bromide stained Agarose
gel with PCR products of Alpha gene using control gene LIS. Lanes
1,2,4,5 shows one alpha gene deletion, lanes 3 & 6 show 2 alpha
gene deletion whereas lane 7 shows normal alpha genes [No alpha gene
deletion]. |
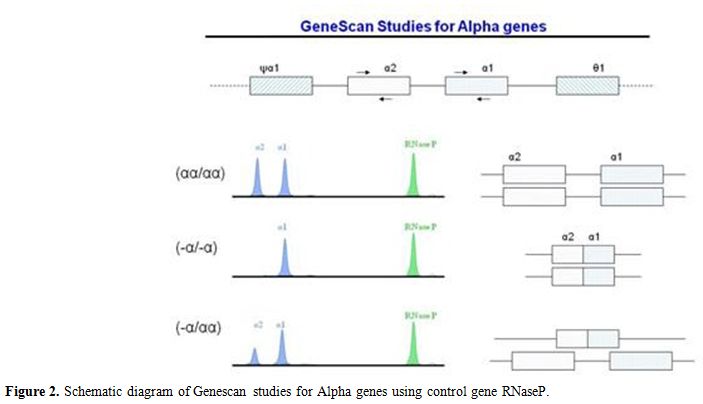 |
Figure 2. Schematic diagram of Genescan studies for Alpha genes using control gene RNaseP. |
Table 2b
shows a correlation between Hb Bart’s at birth and results from
molecular DNA studies. All subjects with two equal peaks on Genescan
showed normal genotype (αα/αα; n=30) and had Hb Bart’s less than 1%.
However, in subjects with two unequal peaks (n=20), GAP PCR confirmed
single deletional alpha thalassaemia (αα/-α3.7)
in all the cases. In one subject with Hb Bart’s of 4.2%, an additional
non-deletional mutation was found by complete alpha gene sequencing
explaining the higher than expected Hb Bart’s. Lastly, in subjects with
one peak on Genescan (n=22), GAP PCR confirmed two gene deletional
alpha thalassaemia (-α3.7/-α3.7) in all the cases.
Table 3
shows the comparative analysis of red cell indices and hemoglobin
values on electrophoresis in the Group C with heterozygous structural
β-globin gene defect (HbS, HbD, HbE & HbC) at birth and at the
follow-up study (n=67). The earlier diagnosis by HPLC was re-confirmed
in all 67 cases by a repeat HPLC and appropriate DNA studies to confirm
the underlying β-hemoglobin variant mutation.
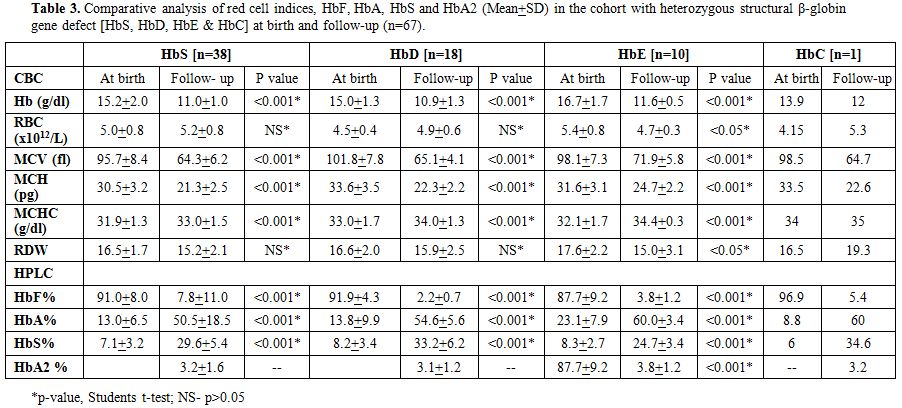 |
Table 3. Comparative
analysis of red cell indices, HbF, HbA, HbS and HbA2 (Mean+SD) in the
cohort with heterozygous structural β-globin gene defect [HbS, HbD, HbE
& HbC] at birth and follow-up (n=67). |
Table 4 shows
the comparative analysis of red cell indices and hemoglobin values on
electrophoresis in Group C (n=41) with low HbA at birth (<10%) and
at the follow-up study (n=29). These babies with Hb A < 10% on HPLC
had no other abnormalities on the HPLC at birth. However, repeat HPLC
in the follow-up study showed that amongst those 41 cases only 29 had
an elevated HbA2 with the mean HbA2 5.0 (range 3.6- 6.2). Sequence
analysis of the entire β-globin gene and the β-globin
gene promoter region using appropriate primers showed that these 29
subjects were carriers for an underlying beta thalassaemia mutation as
described in the earlier study.[11] Repeat red cell
indices at follow- up (n=29) showed that the hemoglobin, RBC counts
with MCHC were normal for the age of the subjects. However, in
comparison to cord blood results, there was a significant drop in
hemoglobin (p < 0.01), MCV (p < 0.0001) and MCH (p < 0.0001)
and a rise in the RBC counts (p <0.01). There was no significant
alteration in MCHC. HbA2 in all these 29 subjects was >3.5%
consistent with a diagnosis of beta thalassaemia trait.
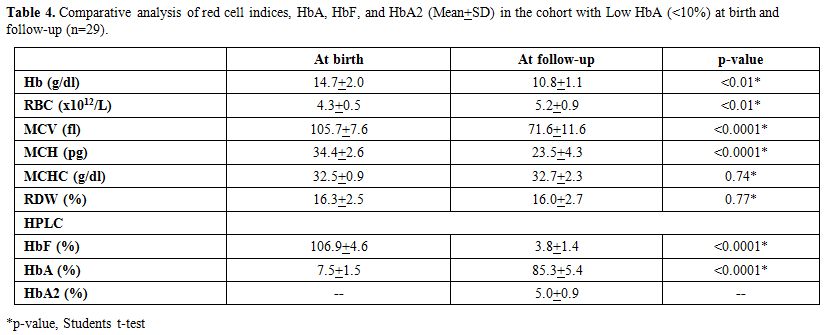 |
Table 4. Comparative
analysis of red cell indices, HbA, HbF, and HbA2 (Mean+SD) in the
cohort with Low HbA (<10%) at birth and follow-up (n=29). |
Discussion
Hemoglobinopathies are quite common in ethnically diverse Omani subjects and represent a major public health concern.[11]
In this context, disease-oriented specific prevention and control
programs are essential and particularly relevant in the context of high
consanguinity rate in this population.[14] In
developed countries, newborn screening accompanied by a continuous,
comprehensive care program (CCCP) has significantly reduced the
morbidity and mortality rate of SCD.[6] Alkindi et al.[11,15]
found that 48.5% subjects showed the presence of Hb Bart’s, and 9.5% of
the same subjects showed the presence of one of the β- hemoglobin
variants namely HbS, HbD, HbE, HbC and beta thalassaemia, although no
case of HbH was detected.[11]
In this follow-up
study, 290 cases from the original neonatal screening study cohort
could be recalled to perform validation using HPLC
and molecular confirmation. Amongst the 31 neonates who on the
HPLC at birth, did not show any HbA (Group A), five were confirmed as
beta thalassaemia major by subsequent HPLC as well as molecular
studies. The remaining twenty-six were confirmed as homozygous sickle
cell disease subjects by molecular studies. The carrier status in the
parents of these 31 subjects was also confirmed, and they were given
appropriate counseling. All these children with SCD were referred to
the paediatric unit for the implementation of CCCP as the most critical
aspect of CCCP is optimizing management by early identification of
affected patients, before the onset of signs and symptoms of
disease. CCCP caregivers also provide extensive parental education
including the prescription of prophylactic penicillin, leading to a
substantial reduction in morbidity and mortality in early childhood.
Oral penicillin prophylaxis in children with sickle cell disease
provides an impressive 85% reduction in
the incidence of infection.[16] However, of concern is that there is a 30% fatality rate observed among children with SCD who develop sepsis.[6,16]
Because infants with sickle cell disease may develop sepsis as young as
four months of age, it is imperative that newborn screening is
universally implemented. The five homozygous thalassaemia children are
currently receiving regular monthly blood transfusions and chelation
therapy.
α-Thalassaemia,
the most common genetic disorder occurs widely throughout Africa, the
Mediterranean countries, the Middle East and the Southeast Asia.[17-19] It is reported that about 45-65% of the ethnic population in the Sultanate of Oman have α-thalassaemia.[11,20] Type 1 and 2 α-thalassaemia are the commonest α- thalassaemias seen. They are caused by partial (type-2;- α) or total (type-1;--) α-gene deletion, which can give rise to various degrees of impaired (-α/αα.--/αα,--/-α) or even completely absent (--/--) hemoglobin α-chain synthesis as well as abnormally low red cell indices (MCV, MCH, MCHC) as seen in our cohort of subjects (Tables 1 and 2).
Amongst
the other 259 subjects that we were able to recall, 151 subjects at
birth had shown the presence of Hb Bart’s by the β-thalassemia short
program (Biorad Variant II) (Group B). The presence of Hb Bart’s in at
birth should lead to investigate the possibility of an underlying alpha
thalassaemia. The mean hemoglobin in this cohort was 11.3 g/dl (Range
7.8-13.4), mean MCV was 67.9 (Range 52.3-77.7). MCV followed by MCH
were the most important discriminators reflecting the microcytic
hypochromic red cell maturation. MCV was the most significant
discriminator (p <0.89E-21; Table 1b).
We
had data on Hb Bart’s quantitation at birth in only 72 subjects of the
151 subjects from group B in whom Hb Bart’s was detected at birth by Hb
Bart’s quantitation using α alpha thalassaemia short program (Biorad Variant II) (Tables 2a and 2b).
These 72 subjects were further subdivided using Hb Bart’s quantitation
and Genescan. In 22 subjects Genescan showed a single peak implying an
underlying deletion of two alpha genes (Figure 2).
Using a cut-off of Hb Bart’s >3% and >2% at birth yielded a
sensitivity of 90.9% and 95.45% respectively. Further, there was a good
correlation with the Hb Bart’s detected at birth with molecular
validation by GAP PCR performed in the follow-up study. In the
remaining 50 subjects who showed two peaks on Genescan, 30 had the two
peaks of equal heights implying the presence of 4 normal alpha genes
which was confirmed by GAP PCR (Figure 1).
These patients had Hb Bart’s at birth, but it was below 1% by Hb Bart’s
quantitation. Thus, saying that Hb Bart’s is pathognomic of an
underlying alpha thalassaemia is not correct. The remaining 20 subjects
showed two peaks of unequal heights on Genescan implying one alpha gene
deletion (Figure 2). Using a
cut-off of >1% Hb Bart’s at birth yielded a sensitivity of 95%. The
highest Hb Bart’s levels were seen in subjects with two alpha gene
deletions (Table 2b). Further, the hemoglobin, MCV, and MCH also showed an inverse correlation with Hb Bart’s quantitation at birth (Table 2a). Unfortunately, the alpha thalassaemia short program kits have been currently discontinued by Biorad.
Amongst
the remaining hundred and eight subjects, 67 subjects were shown to
have the presence of a beta gene structural variants namely HbS, HbD,
HbC and HbE (Table 3). The mean
abnormal hemoglobin at birth respectively for HbS, HbD, HbE was 7.1%,
8.2 and 8.3% which respectively rose to 29.6% (range 22.4-44.1), 33.2%
(range 28.9-37.4) and 24.7% (range 22.3-27.1) (Table 3).
In the single patient with HbC the abnormal hemoglobin rose from 6% to
34.6% in the follow-up study. Thus, in the follow-up study, there was a
100% concordance and validation (including molecular confirmation) with
the observations made at birth with regards to beta-globin structural
variants.
In the remaining 41 subjects with HbA levels below 10%
at birth, it is likely that beta thalassaemia minor was a possibility,
as in the absence of HbA2 HPLC cannot show diagnostic discrimination in
such cases at birth. Repeat HPLC in the follow-up study showed that
amongst these 41 cases only 29 had an elevated HbA2 with the mean HbA2
5.0 (range 3.6-6.2) (Table 4),
and which was confirmed in all these 29 cases by sequencing the β
globin gene including the promoter region, all exons and introns. The
mutation found were consistent with the earlier report.[11]
Using a cut-off of HbA <10 at birth yielded a sensitivity of only
67%, that improved to 90% with a cutoff of HbA <9 at birth and to
100% with a cut-off of HbA <8.0 at birth.
The study has certain
limitations, especially regarding the small sample size. Although the
original study cohort included 3740 newborn babies, we were able to
recruit only 290 subjects (7.75%) mainly due to logistical reasons.
Although there were no cases of HbH (3 genes abnormalities;(--/-α)
or Hydrops fetalis (4 genes abnormalities; (--/--)) in this study,
cases with HbH have been reported in other studies from Oman.[12] Furthermore, we also did not encounter the single gene deletional defect (αα/-α4.2) in the follow-up subjects although it had been reported in the initial study.[11]
Conclusions
Although
the study population is small, what needs to be highlighted is that in
this small cohort of subjects, HPLC and CBC were instrumental in the
initial approach to identifying an underlying haemoglobinopathy. This
follow-up study found 100% concordance with the results of initial HPLC
and CBC results and was this was confirmed with appropriate molecular
studies. Hb Bart's ≥ 2% yielded 95.45% sensitivity to identify two
α-globin gene deletions, whereas Hb Bart's ≥ 1% yielded 95% sensitivity
to identify one α-globin gene deletions. A cut-off of HbA <10 at birth yielded a sensitivity of only 67%, which improved to 90% and 100% with a cutoff of HbA <9 and <8.0 at birth respectively.
Furthermore,
the hemoglobin, MCV, and MCH, correlated well with the alpha genotype
by GAP PCR and the Hb Bart’s level by the quantitation method. Also,
MCV ≤ 95 fl and MCH ≤ 30 pg yielded 100% sensitivity to identify two
α-globin gene deletions. Considering the simplicity, consistency and
rapid results, HPLC can be a major tool in our neonatal screening
program.
Acknowledgements
We
wish to thank the hospital administration for the use of hospital
material in this study. The initial study was supported by a grant from
His Majesty’s Research Project IG/MED/HEAM/05/01.
References
- Leikin SL, Gallagher D, Kinney TR, Sloane D, Klug
P, Rida W. Mortality in children and adolescents with sickle cell
disease. Cooperative Study of Sickle Cell Disease. Pediatrics. 1989;
84:500-8. PMID:2671914. PMid:2671914
- Serjeant
GR. Natural history and determinants of clinical severity of sickle
cell disease. Curr Opin Hematol. 1995; 2:103-8. PMID: 9371979 https://doi.org/10.1097/00062752-199502020-00001 PMid:9371979
- Koko
J, Dufillot D, M'Ba-Meyo J, Gahouma D, Kani F. Mortality of children
with sickle cell disease in a pediatric department in Central Africa.
Arch Pediatr. 1998; 5:965-9 https://doi.org/10.1016/S0929-693X(98)80003-1 PMID:9789626
- Thomas
C, Lemerle S, Bernaudin F, Feingold J, Guillou-Bataille M, Reinert P.
Sickle cell anemia: study of the pediatric mortality in Ile de France
from 1985 to 1992. Arch Pediatr. 1996; 3:445-51. https://doi.org/10.1016/0929-693X(96)86402-5 PMID:8763714
- Platt
OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al.
Mortality in sickle cell disease. Life expectancy and risk factors for
early death. N Engl J Med. 1994; 330:1639-44 https://doi.org/10.1056/NEJM199406093302303
- Quinn
CT. Sickle cell disease in childhood: from newborn screening through
transition to adult medical care. Pediatr Clin North Am. 2013
Dec;60(6):1363-81. https://doi.org/10.1016/j.pcl.2013.09.006
- Riddington
C, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal
infection in children with sickle cell disease. Cochrane Database Syst
Rev.;2002, (3):CD003427. https://doi.org/10.1002/14651858.CD00342
- Buchanan
GR, Smith SJ. Pneumococcal septicemia despite pneumococcal vaccine and
prescription of penicillin prophylaxis in children with sickle cell
anemia. Am J Dis Child,1986, 140:428–432. PMID:3962935. PMid:3962935
- Vichinsky
E, Hurst D, Earles A, Kleman K, Lubin B Newborn screening for sickle
cell disease: effect on mortality. Pediatrics.;1988, 81(6):749-55.
PMID:3368274. PMid:3368274
- King L,
Fraser R, Forbes M, Grindley M, Ali S, Reid M. Newborn sickle cell
disease screening: the Jamaican (1995-2006). J Med Screen.
2007;14(3):117-22. https://doi.org/10.1258/096914107782066185
- Alkindi
S, Al Zadjali S, Al Madhani A, Daar S, Al Haddabi H, Al Abri Q, et al.
Forecasting hemoglobinopathy burden through neonatal screening in Omani
neonates. Hemoglobin.2010 Jan;34(2):135-44. https://doi.org/10.3109/03630261003677213
- Alkindi
SS, Alzadjali S, Daar S, Sindhuvi E, Wali Y, Pathare AV, et al. A
stepwise α-thalassemia screening strategy in high-prevalence areas.
experience (1995-2006). Eur J Haematol. 2013 Aug;91(2):164-9. https://doi.org/10.1111/ejh.12136
- Jassim
N, Al-Arrayed S, Gerard N, Al-Mukharraq H, Al-Ajami A, Ramasawmy R, et
al. A mismatched-primer polymerase chain reaction- restriction fragment
length polymorphism strategy for rapid screening of the polyadenylation
signal mutation alpha(T-Saudi) (AATAAA-->AATAAG) in the
alpha2-globin gene. Hemoglobin. 1999 Aug;23(3):213-20. https://doi.org/10.3109/03630269909005701 PMid:10490133
- Rajab A, Patton MA. A study of consanguinity in the Sultanate of Oman. Ann Hum Biol. 2000 May-Jun;27(3):321-6. PMID: 10834296. https://doi.org/10.1080/030144600282208 PMid:10834296
- Alkindi
S, Pathare A, Al-Madhani A, Al-Zadjali S, Al-Haddabi H, Al- Abri Q, et
al. Neonatal Screening: Mean haemoglobin and red cell indices in cord
blood from Omani neonates. Sultan Qaboos Univ Med J.2011, 11(4):462-9.
PMid:22087394 PMCid:PMC3206748
- Piel FB,
Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle
cell anaemia in children under five, 2010-2050: modelling based on
demographics, excess mortality, and interventions. PLoS Med.
2013;10(7): e1001484 https://doi.org/10.1371/journal.pmed.1001484
- Higgs
DR, Vickers MA, Wilkie AO, Pretorius IM, Jarman AP, Weatherall DJ.: A
review of the molecular genetics of the human a-globulin gene cluster.
Blood 1989; 73(5):1091-1108. PMID:2649166.
- Weatheral DJ. The thalassaemias. In: Beutler E, Litchmen MA, Coller BS, Kipps TJ, editors. William's Hematology. 5th Edition (International edition). New York: McGraw-Hill Inc, Health Professions Division, 1995:581-615.
- Nasserullah
Z, Al-Jame A, Abu Srair H, Al Qatari G, Al Naim S, Al Aqib A et al.
Neonatal screening for sickle cell disease, glucose-6- phosphate
dehydrogenase deficiency and α-thalassaemia in Qatif and Al Hassa. Ann
Saudi Med:1998; 18(4):289-92. https://doi.org/10.5144/0256-4947.1998.289 PMid:17344674
- White
JM, Christie BS, Nam D, Daar S, Higgs DR. Frequency and clinical
significance of erythrocyte genetic abnormalities in Omanis. J Med.
Genet. 1993; 30(5):396-400. https://doi.org/10.1136/jmg.30.5.396 PMid:8320702 PMCid:PMC1016376
[TOP]