Raffaele Manna1,2 and Donato Rigante3,2..
1 Institute of Internal Medicine, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
2 Periodic Fevers Research Centre, Università Cattolica Sacro Cuore, Rome, Italy.
3 Institute of Pediatrics, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
Correspondence to: Raffaele Manna, MD, PhD, Institute of Internal
Medicine, Fondazione Policlinico Universitario A. Gemelli IRCCS,
Università Cattolica Sacro Cuore, Rome, Largo A. Gemelli 8, 00168 Rome,
Italy. Tel. +39 06 30159433. Fax. +39 06 35502775. E-mail:
raffaele.manna@policlinicogemelli.it
Published: May 1, 2019
Received: January 14, 2019
Accepted: March 7, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019027 DOI
10.4084/MJHID.2019.027
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Recurrent
self-limited attacks of fever and short-lived inflammation in the
serosal membranes, joints, and skin are the leading features of
familial Mediterranean fever (FMF), the most common autoinflammatory
disorder in the world, transmitted as autosomal recessive trait caused
by MEFV gene mutations. Their
consequence is an abnormal function of pyrin, a natural repressor of
inflammation, apoptosis, and release of cytokines. FMF-related mutant
pyrins are hypophosphorylated following RhoA GTPases’ impaired activity
and show a propensity to relapsing uncontrolled systemic inflammation
with inappropriate response to inflammatory stimuli and leukocyte
spread to serosal membranes, joints or skin. Typical FMF phenotype 1
consists of brief episodes of inflammation and serositis, synovitis,
and/or erysipelas-like eruption, whereas phenotype 2 is defined by
reactive amyloid-associated (AA) amyloidosis, which is the most ominous
complication of FMF, in otherwise asymptomatic individuals.
Furthermore, FMF phenotype 3 is referred to the presence of two MEFV
mutations with neither clinical signs of FMF nor AA amyloidosis. The
influence of epigenetic and/or environmental factors can contribute to
the variable penetrance and phenotypic heterogeneity of FMF.
Colchicine, a tricyclic alkaloid with anti-microtubule and
anti-inflammatory properties, is the bedrock of FMF management: daily
administration of colchicine prevents the recurrence of FMF attacks and
the development of secondary AA amyloidosis. Many recent studies have
also shown that anti-interleukin-1 treatment is the best therapeutic
option for FMF patients nonresponsive or intolerant to colchicine. This
review aims to catch readers’ attention to the clinical diversity of
phenotypes, differential diagnosis, and management of patients with FMF.
|
Introduction
Primary
dysfunction of the innate immune system is the architrave of
autoinflammatory disorders, in which there is no evidence of adaptive
immunity involvement, neither high-titer autoantibodies nor
antigen-specific T cells, and no infectious triggers.[1] People with
hereditary autoinflammatory disorders display periodically-recurring
clinical features consisting of fever and inflammation with
symptom-free intervals of different duration between febrile
attacks.[2] In this group of diseases, familial Mediterranean fever
(FMF) occupies a primary seat, as it is the most common
autoinflammatory disorder worldwide,[3] although it has been
historically associated with people living around the Mediterranean
basin as Turks, Arabs, Sephardic Jews, and Armenians.[4]
The Ancient Heredity of Familial Mediterranean Fever
In
1945 the allergologist Sheppard Siegal described as “benign paroxysmal
peritonitis” his own disease, which was similar to the characteristics
presented by other five Jewish patients: they all had cutaneous signs,
recurrent peritonitis, and periodic fever attacks.[5] In 1954 Hobart
Reimann was the first clinician who talked about “FMF,” defining its
“periodic” outstanding features, noting that the adjectives cyclic, rhythmic, episodic, relapsing, recurrent, paroxysmal and intermittent
had been used interchangeably to highlight the disease.[6] In 1955
professor Harry Heller and his colleagues described the overall FMF
clinical picture in detail, studied its inheritance, and found
FMF-associated nephropathy deriving from amyloidosis as an ominous
long-term complication of FMF.[7] FMF was the first autoinflammatory
disorder for which the causing gene was identified in 1997 by two
independent groups, American and French.[8-10] The gene was called MEFV (from Mediterranean FeVer)
and is located on the short arm of chromosome 16 (p13.3) between the
genes linked to polycystic kidney disease and Rubinstein-Taybi
syndrome, spanning approximately 14 Kb of genomic DNA and comprising 10
exons: it encodes a 781 amino acid-protein with a molecular weight of
86 kD which was called “pyrin” (meaning “fever” in Greek) by the
American group and “marenostrin” (using the ancient Latin name “mare
nostrum” for the Mediterranean sea) by the French one.[11] This
discovery represented the starting point to understand the pathogenesis
not only for FMF but also for other autoinflammatory disorders. During
the last years, there have been lots of studies demonstrating a growing
interest of the scientific community in the search for autoinflammatory
mechanisms in many other inflammatory conditions.[12,13]
The Size of a Borderless Disease
Although
genetic research into FMF began around 1997, we still lack a complete
picture of its genetic variation, carrier frequency, and
penetrance.[14] Ethnic distribution of peculiar MEFV
variants was initially considered a feature of FMF, and the disease was
mostly recognized in people living around the Mediterranean basin and
deriving from the ancient Mesopotamia or Phoenician lands: Armenians,
non-Ashkenazi Jews (above all Sephardic Jews, who emigrated to the
Iberian peninsula, in fact, Sephardi
means “Spain” in Hebrew), Turks, Arabs (mostly North-Africans such as
Maghrebins) and Druze population (an ethnoreligious Arabic-speaking
group originating in Western Asia). The clinical diagnosis of FMF among
these populations is enhanced by the widely-recognized higher
prevalence of the disease. In addition, it is unclear whether all MEFV
variants are true disease-causing mutations, as only five founder
mutations (V726A, M694V, M694I, M680I, and E148Q) have been related to
74% of typical FMF cases in Armenians, Arabs, Jews and Turks.[15]
However, because of so many migrations during the past centuries, the
gene causing FMF has also been spread to Western Europe as well as to
the Americas, China, Japan, Australia and New Zealand.[16] The higher
frequencies of MEFV variants in some populations could be explained by an increased resistance against specific pathogens, such as Yersinia pestis or intracellular bacteria such as Mycobacterium tuberculosis,
or by raised protection from asthma.[17] In Italy, FMF is erroneously
considered a rare disease (“rare” means that its prevalence is less
than 1 per 2000 inhabitants): various historical reasons account for
the presence of the FMF gene in Italy, as Greek colonization of Sicily
and Southern Italian regions in the VIII century B.C., the arrival of
the first Christians in Rome under the Roman Empire in the I-II century
A.D., and the Arab conquest of Sicily in the IX century A.D.[18,19] The
observation of the same mutations and haplotypes in populations that
have been isolated for centuries with high rates of consanguinity
indicates that most cases of FMF are descended from a very ancient pool
of founders.
Pyrin, an Intracellular Sentinel against Infections
Before MEFV
discovery, it was commonly known that FMF attacks were characterized by
a massive sterile flux of polymorphonuclear leukocytes into some body
regions, which were serosal membranes, joints, and skin.[20,21] A huge
number of studies dedicated to pyrin is revealing new molecular details
about the general processes of cellular defense against infections,
inflammation and apoptosis. Indeed, pyrin is largely expressed in the
white blood cells and has a pivotal role in the activation of
inflammasome and processing of the potent pyrogenic cytokine
interleukin (IL)-1ß: in fact, it is involved in the activation of
caspase-1 and release of active IL-1, as a structural part of the
inflammasome complex.[22] In particular, the “pyrin domain” of this
protein is a member of the death-domain-fold superfamily and is
involved in the apoptotic pathway modulation through caspase
recruitment and production of IL-1.[23] IL-1 transcriptional pathways
are also misregulated in FMF attack-free periods, supporting the
presence of subclinical inflammation between attacks, though different
studies have not confirmed this hypothesis.[24,25] Pyrin activity is at
the level of cytoskeletal assembly, and specific microtubule assembly
inhibitors prevent pyrin-mediated caspase-1 activation and secretion of
IL-1 in peripheral blood mononuclear cells of FMF patients.[26] Recent
experimental studies have suggested that pyrin function is mediated by
RhoA (Ras homolog gene family-member A) GTPases, enzymes targeted to
the plasma membrane by the addition of a geranylgeranyl lipid
tail: more specifically, FMF mutations in the pyrin B30.2 domain
decrease the threshold of activation of the pyrin inflammasome,
generally activated by various RhoA-inhibiting toxins produced by both
Gram-negative and Gram-positive bacteria. In a normal condition, the
phosphorylated pyrin binds to regulatory proteins that block the pyrin
inflammasome, but inhibition of RhoA effector kinases decreases the
phosphorylation of pyrin, which in turn activates pyrin
inflammasome.[27] Triggers stimulating at a cellular level the periodic
occurrence of acute clinical attacks of FMF are not known, even if
different chronic infections such as Helicobacter pylori
infection or small bowel bacterial overgrowth might tune patients’
inflammatory responses.[28,29] Other current studies suggest that non-MEFV
genetic systems, epigenetic, and environmental modifiers might
interplay with pyrin, influencing the clinical expression of FMF.
Colchicine, a major neutral alkaloid from Colchicum
species, suppresses pyrin inflammasome activity through direct
activation of RhoA, disabling host cell cytoskeletal organization and
causing an anti-chemotactic effect on the polymorphonuclear cells.[30]
A Host of Genotype Studies
FMF
is inherited in a recessive manner, although recent studies have
suggested that some heterozygotes manifest a spectrum of findings from
classic FMF to mild FMF.[31] The most frequent MEFV mutations are contained in the exons 10 and 2 and are heterogeneously distributed in different populations.[32]
Four mutations represent more than 70% of the mutated alleles in FMF
cases of Mediterranean ancestry: M694V, M694I, M680I and V726A, all in
the exon 10. Genotypes including two mutations located within
mutational MEFV “hot-spots” (codons 680 or 694) in the exon 10 have been associated with the most severe phenotypes of FMF.[33]
An unsolved issue is the possibility of genotype-phenotype
correlations: for instance, M694V homozygosis, M680I homozygosis and
heterozygosis for M608I/M694V genotypes are usually associated with
severe clinical pictures and more frequent incidence of amyloidosis,
while V726A is associated with milder disease and less frequent
amyloidosis.[34] Understanding the correlation
between FMF phenotype and genotype is further obscured by the existence
of complex alleles, modifier loci, and potential epigenetic factors. In
the United States FMF is frequently encountered in Ashkenazi Jews and
immigrants from the Middle East and Armenia. In Germany, most FMF
patients are of Turkish origin. In France, there is a relatively large
FMF population that originated from North Africa. Among Armenians, the
carrier rate for FMF is approximately 1:7 with a disease rate of
roughly 1:500.[35] The E148Q is the least penetrant
mutation of FMF, frequently found in the less severe pictures so that
it has been considered a polymorphism, not a true disease-causing
mutation.[36] The R202Q mutation is also considered a genetic polymorphism, present in about 15% of unaffected populations.[37]
Some genes have been tested to assess their possible modifying effect
on the clinical features of FMF: for instance, the alpha/alpha genotype
of the serum amyloid-A gene (SAA1) is associated with increased risk of amyloidosis in FMF patients, especially if homozygous for the M694V mutation.[38] Moreover, Berkun et al. showed that FMF patients carrying mutations in the NOD2/CARD15
gene, which is involved in Blau syndrome, an autoinflammatory disorder
starting in the first years of life with noncaseating epithelioid
granulomas affecting joints, skin and eye, variably associated with
heterogeneous systemic features,[39] are prone to a more severe FMF course and higher development of colchicine resistance.[40] A Kaleidoscopic Disease with Protean Faces
FMF
is characterized by short-lived and self-resolving attacks of fever,
abdominal, thoracic or joint pain and systemic inflammation which recur
over time combined with intercritical periods of apparent wellness:[41] both clinical features and disease severity may vary among different ethnic groups.[42]
General triggers of FMF attacks may be emotional stress, strenuous
physical activity, menses, nutritional changes, use of contraceptives,
hypovitaminosis D, etc. Bouts of fever with painful symptoms related to
the abdomen, chest or one or multiple joints, singularly or in various
combinations, are the most frequently declared symptoms by people
living throughout many areas of the Mediterranean basin. Fever is
present in 96% of inflammatory episodes: body temperature can peak over
40°C; it usually appears suddenly and lasts from 6 to 72 hours,
preceded by chills in about ¼ of cases. Two phenotypically independent
pictures of FMF can be recognized: [a] acute short-lasting painful
febrile attacks of peritonitis, pleuritis, or arthritis (phenotype 1),
and [b] nephropathic amyloidosis (phenotype 2), which can lead to
terminal renal failure even at a young age, and no other clinical
symptom. There is a third picture (phenotype 3) characterized by two MEFV mutations with neither clinical signs of FMF nor of amyloid-associated (AA) amyloidosis.[43]
Attacks do not have a regular periodism: their frequency varies from
once per week to once per decade. Over the course of this lifelong
illness, an affected individual will probably have several forms of
febrile and painful episodes, but the recurrence of one type over many
years is most frequent.[44] Onset is usually during
the first decade in about 50% of cases, and symptoms might also start
in the first months of life. As clinical manifestations of FMF appear
quite early; they might be confused with a variety of diseases
occurring in the pediatric age: for instance, recurrent febrile
tonsillitis may be a symptom of FMF attacks, especially in early
childhood,[45] entering in a challenging differential
diagnosis with periodic fever, aphthous stomatitis, pharyngitis, and
adenitis (PFAPA) syndrome, a typical recurrent disorder occurring in
children less than 5 years, but also reported in adults.[46-49]Abdominal
pain, due to peritoneum inflammation resembling acute abdominal disease
like appendicitis, cholecystitis, ureteral stones or pelvic
inflammatory disease, is present in about 90% of patients and is
frequently associated with small amounts of ascites upon ultrasound
investigation. Peritoneum inflammation may also occur in the form of
massive ascites with myofibroblast proliferation in the mesenteric
region as an initial presentation of FMF.[50] Sometimes (in 30-40% of cases) patients might undergo unnecessary surgery.[51]
Unilateral or bilateral pleural effusion can be demonstrated in 50-60%
of patients and is characterized by quick resolution, while recurrent
pericarditis can be observed in a small percentage of FMF patients
(1-2.5%) during febrile attacks.[52,53] Intense
scrotal pain simulating a testicular torsion is frequent in children
with FMF, but it usually requires a conservative management.[54]
Joint involvement is reported in 45% of cases and might present as
transient arthralgia or oligoarthritis: short-lasting attacks begin
without prodromes, involve large joints and suddenly disappear after
24-48 hours with no sequelae. Rarely arthritis may last for more than
one week or even develop destructive features.[55]
Some patients have painful, swollen and self-limited erythematous skin
lesions on the legs: these lesions resemble a skin infection called
erysipelas and are quite specific for FMF.[56]
Another symptom associated with FMF is muscle pain, which might be
severe and paralyzing: this muscular involvement is not prevented by
colchicine and does not respond to nonsteroidal anti-inflammatory
drugs, but only to corticosteroids. Muscular manifestations can present
with severe febrile myalgia having three different patterns:
spontaneous, effort-induced (i.e., exertional leg pain) and prolonged
with an overall duration of 6 weeks.[57,58] In recent times the clinical spectrum of FMF has expanded and many non-canonical manifestations have been reported.[59]
For instance, neurologic signs other than constitutional headache have
been described in some FMF patients, such as recurrent aseptic
meningitis, demyelinating disorders, recurrent peripheral facial palsy
and even stroke.[60]Amyloidosis
is the most dreadful complication of late-diagnosed, untreated or
neglected FMF, which results from the deposition in different organs of
a fatty-like substance, which is a cleavage product of serum amyloid-A,
an acute-phase reactant produced by the liver. Main organs involved by
amyloid deposition are kidney, gut, spleen, liver, heart and endocrine
glands. Renal amyloidosis culminating in renal failure, which occurs in
as many as 60% of untreated patients with FMF, is the major cause of
death in FMF. Recent studies have focused on the polymorphism of the
SAA1 gene as a genetic contributor to the development of
amyloidogenesis, but found no influence by the major histocompatibility
complex.[61] The pathogenic role of environmental
factors in FMF-related amyloidosis such as the country of origin is
suggested by the lower incidence of amyloidosis in Jews living outside
the Mediterranean basin: for instance, Armenians living in Armenia have
a much higher incidence of amyloidosis than Armenian Americans, even
before the introduction of colchicine. In addition, amyloidosis appears
to be less common among Iraqis, Ashkenazi Jews and Arabs.[62] The association between amyloidosis and the mutation M694V is widely reported,[63]
but non-amyloid glomerulopathies such as IgM or IgA nephropathy, focal
and diffuse proliferative glomerulonephritis, and rapidly progressive
glomerulonephritis have also been observed. Also, some
non-granulomatous vasculitides, such as Henoch-Schönlein purpura (in
2.6-5% of cases), polyarteritis nodosa (0.8-1%) and Behçet’s disease
(0.5%) have been associated with FMF.[64-66] Over the years, an increased rate of MEFV
carriers has also been found in complex multifactorial diseases such as
ankylosing spondylitis and multiple sclerosis, implicating this gene
and its pathways in the development of such disorders.[67]
A few patients with FMF might also have attacks characterized by
macrophage activation syndrome, in which well-differentiated
mononuclear cells exhibiting hemophagocytic activity have swarmed
different organs and systems, giving rise to a dramatic clinical
picture consisting of persistent fever, multi-organ damage, cytopenia,
hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia with
high mortality rates.[68,69] Lastly, a retrospective
study related to over 8.000 Jewish patients with FMF registered at the
Tel Hashomer Hospital (with a mean age of 43.74 ± 14.7 years) revealed
that there is a significantly lower incidence of cancer in FMF patients
than in the general population of Israel, and which might be attributed
to a direct physiologic effect of FMF or the antimitotic effects of
colchicine administered on the long run.[70] Table 1 shows the main clinical features reported by 373 patients managed in the Periodic Fevers Research Center of our University.
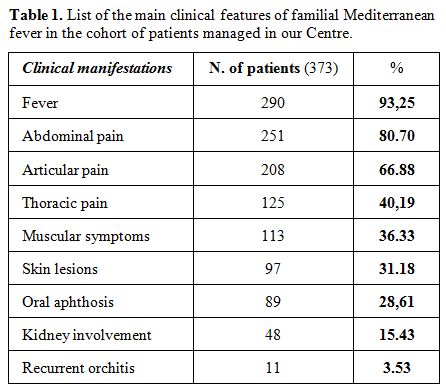 |
Table
1. List of the main clinical features of familial Mediterranean fever in the cohort of patients managed in our Centre. |
Searching for Universal Diagnostic Criteria of Familial Mediterranean Fever
Traditionally,
diagnosis of FMF has been based on the typical clinical manifestations
and physician's insight, though a purely clinical diagnosis is
complicated if there are febrile episodes without serositis or if
amyloidosis is initially demonstrated without a clear history of fever
and serositis. There is no specific test for FMF diagnosis, and some
diagnostic criteria have been suggested, all based on clinical data and
supported by the patient’s ethnic origin or family history. During past
years many authors have tried to develop different diagnostic
flow-charts and several sets of criteria have been proposed since 1967,
when Sohar et al. deduced that periodically-recurrent fevers and
self-limited painful manifestations in various sites (abdomen, chest,
joints, or skin), not explained by any other causative factor, were a
mandatory FMF feature. They also recognized the importance of AA
amyloidosis and the patient’s origin from the areas around the
Mediterranean sea. The Tel Hashomer Hospital criteria by Sohar’s group
(listed in Table 2) were
promulgated starting from clinical observations in adult Israeli
population, and are the most widely used for diagnosis of FMF in most
clinical settings. If 2 major Tel Hashomer criteria or 1 major
criterion and 2 minor ones are satisfied the diagnosis of FMF can be
confirmed.[71]
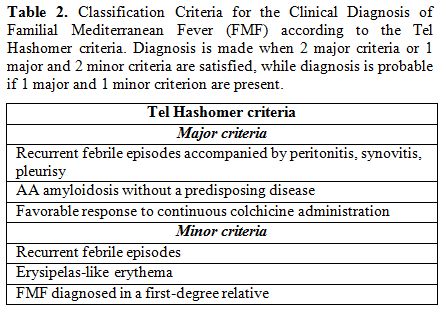 |
Table
2. Classification Criteria for the Clinical Diagnosis of Familial
Mediterranean Fever (FMF) according to the Tel Hashomer criteria.
Diagnosis is made when 2 major criteria or 1 major and 2 minor criteria
are satisfied, while diagnosis is probable if 1 major and 1 minor
criterion are present. |
In
1997 new criteria were validated by Livneh et al. to corroborate the
clinical elements included in the Tel Hashomer criteria, but excluding
other manifestations, like amyloidosis not explained by any other
causative factor, less common at the onset of FMF. Two versions of
these criteria, one more conservative and extensive than the other,
were created, both with a sensitivity and specificity above 95%: the
presence of at least 1 of 4 major criteria related to “typical” disease
attacks or 2 of 5 minor criteria or 1 minor criterion plus 5 of 10
supportive criteria should suggest the diagnosis of FMF (see Table 3).[72]
 |
Table 3. Classification
Criteria for the Clinical Diagnosis of Familial Mediterranean Fever
(FMF) according to Livneh et al. Diagnosis of FMF requires ≥1 major criteria, or ≥ 2 minor criteria, or 1 minor criterion plus ≥5 supportive criteria
(family history of FMF, appropriate ethnic origin, age less than 20
years at disease onset, severity of attacks requiring bed rest,
spontaneous remission of symptoms, presence of symptom-free intervals,
transient elevation of inflammatory markers, episodic proteinuria or
hematuria, nonproductive laparotomy with removal of a “white” appendix,
consanguinity of parents) or 1 minor criterion plus ≥ 4 of the “first”
five supportive criteria.
“Incomplete” attacks are defined as painful and recurrent flares that
differ from typical attacks in 1 or 2 features, as follows: 1) normal
temperature or lower than 38°C; 2) attacks longer than 1 week or
shorter than 6 hours; 3) no signs of peritonitis recorded during acute
abdominal complaint. |
Diagnosis
of FMF in children has been established using the same clinical
criteria created for adults. However, a frequent delay in the
appearance of a complete clinical picture in very young children, the
presence of atypical signs, and absence of a suggestive family history
may cause additional difficulty. Moreover, the evidence that some
criteria were of poor relevance for children with FMF and differences
of some clinical manifestations in younger children (who might have
shorter attacks, infrequent chest pain, or even fever alone) prompted a
Turkish group of clinicians in 2009 to formulate new FMF criteria for
the pediatric population (the so-called Turkish FMF Pediatric
criteria), listed in the Table 4.[73] The pediatric cohorts where these criteria were evaluated included only cases genetically ascertained to have two MEFV mutations, regardless of their phenotypes.
 |
Table 4. Classification
Criteria for the Clinical Diagnosis of Familial Mediterranean Fever
(FMF) according to Yalçinkaya and Ozen (Turkish FMF Pediatric
Criteria). Diagnosis of FMF requires the presence of 2 out of 5
criteria (in Turkish children). |
These
Turkish criteria for the diagnosis of FMF yielded a better sensitivity
in an international cohort of children from either European or Eastern
Mediterranean regions, though their specificity was lower than in
previous criteria.[74] Anyway, all used diagnostic
criteria should need further improvements, such as validation on
broader cohorts of probands, and the inclusion of genetic data.
Federici et al. developed new classification criteria for the diagnosis
of different autoinflammatory disorders through the identification of
‘positive’ and ‘negative’ criteria correlated with each disease,
suggesting that the presence of aphthous stomatitis, urticaria-like
rashes, cervical lymph node enlargement and febrile episodes lasting
more than 6 days should exclude the diagnosis of FMF.[75]There is no specific laboratory examination to support the diagnosis of FMF, except for MEFV
analysis. During typical acute attacks, blood tests show a generalized
increase of the inflammatory parameters (erythrosedimentation rate,
C-reactive protein, serum amyloid-A, immunoglobulins) with a parallel
neutrophil leukocytosis (until and over 20.000/mm3).[76]The
role of genetics in supporting the diagnosis of FMF is essential, but
the genetic analysis should never substitute both clinical process and
clinician’s judgment. Following MEFV
cloning, the genetic test for FMF has become available in many
countries, providing a new exciting tool for diagnostic confirmation.
Genetic diagnosis is definite when two mutations, also nonidentical,
are present in the two MEFV
alleles; if only one or no pathogenic mutation is found the clinical
diagnosis of FMF is still possible due to the potential occurrence of
occult mutations. Of course, for patients with typical clinical
pictures and appropriate ethnic origin, FMF diagnosis can be made
without a genetic confirmation, which is vice versa contributory for
cases with atypical presentation, the absence of family history, or
unusual ethnic origin. In particular, genetic testing might reveal no
known mutation in 30-40% of patients: in this case if clinical
manifestations are convincingly those of FMF the patient is put on an
open trial with colchicine for 3-6 months. If there is a positive
response and symptoms reappear after stopping colchicine, it is assumed
that the patient has FMF.[77]Furthermore,
diagnosis of FMF in very young heterozygous children should be made
with caution, as FMF heterozygous children can present with an FMF-like
disease in early childhood, which does not differ from that seen in
patients carrying two mutated alleles, although it may disappear with
age. Therefore, a careful follow-up of heterozygous children is of
paramount importance before establishing a definite diagnosis.[78]
For individuals with two FMF-related mutations who do not report
symptoms, if there are risk factors for amyloidosis (such as the
country, family history, and persistently elevated inflammatory
markers, particularly serum amyloid-A), a close follow-up should be
started and at least colchicine prophylaxis considered.
Ancient and Modern Treatments
Since
1972 colchicine is the anchor drug used in patients with FMF. Lifelong
prophylaxis with colchicine is safe and effective in preventing attacks
of FMF and also the well-known complication of amyloidosis:[79]
a therapeutic trial with colchicine for at least three-six months is
useful to ascertain whether there is a decrease in the severity or in
the frequency of FMF flares in every patient suspected to have FMF.[80]
Continuative colchicine prophylaxis, even if started during childhood,
has no effects on the normal final expected growth of children. By
contrast, colchicine is not effective if administered during acute
flares of FMF. The minimal daily dose in adults is 1 mg/day, but in
children there is no definitely suggested dose, and management should
be driven by the occurrence of clinical symptoms and inflammation on
laboratory tests. Other factors, such as the genotype or body surface
area may help to manage colchicine dose in a more individualized
fashion.[81] The initial dose in children is usually
0.5-1 mg/day regardless of age and body weight, with little increases
until disease control is achieved. The rate of adverse events for
colchicine, mainly gastrointestinal effects, is low. Neutropenia, hair
loss and allergies are infrequent side effects. A colchicine-related
myopathy combined with the mild-to-severe elevation of creatine kinase
may appear two weeks to several months after initiation of treatment.
Moreover, since in vitro high doses of colchicine stop mitosis and
colchicine displays its effect by fixating the intracellular
microtubules, arresting their polymerization and finally disrupting
mitosis and motility systems within the cells, in theory, this drug
could interfere with patients’ fertility, though many studies have
proved no clearly adverse effects on gametogenesis.[82]
However, if untreated, FMF itself can lead to amyloid deposits in the
testis and ovary, resulting in infertility: therefore, patients with
FMF may safely continue to use colchicine throughout the reproductive
phase of their life.[83]Complete
remission of FMF under colchicine is achieved in 65% of patients and
partial remission in 30%, while around 5% of patients remain
nonresponsive.[84] True resistance to colchicine is a rare event, mostly observed in patients displaying peculiar MEFV
genotypes and occurring despite the regular use of colchicine at the
maximal doses. Low adherence to colchicine administration may be a key
component of resistance and should be assessed.[85]
Since long-term colchicine prophylaxis may be complicated by
gastrointestinal side effects, by our experience, we usually recommend
lactose-free diet and treatment of intestinal bacterial overgrowth to
improve colchicine tolerance.[86] There are also FMF
patients with low disease activity who might become utterly free of
attacks for a long time and even stop colchicine prophylaxis: long-term
remission of FMF, characterized by a time interval of at least 3 years
without FMF clinical manifestations and off-colchicine, after a period
of FMF activity, is rather infrequent. The prevalence of disease
remission in the FMF population is estimated at 3.3%, based on a study
by Ben-Zvi et al. from the largest center for diagnosis and treatment
of FMF in Israel.[87] These patients seem to have
distinct clinical, demographic and molecular characteristics,
allocating them to the mildest end of the disease severity spectrum of
FMF. This phenotype is comparable to that of patients with late-onset
FMF.[88]FMF
protracted arthritis, mostly affecting hip or knees, has shown results
following treatment with tumor necrosis factor inhibitors.[89]
Characterization and early identification of FMF patients with
uncontrolled inflammatory activity have become more important after the
availability of new treatment options for the prevention of
disease-associated complications and permanent damages of FMF. After
the demonstration of inflammasome dysregulation as the dominating
pathogenetic mechanism in different autoinflammatory disorders
including FMF, IL-1β has been shown as the most intriguing target to
attack.[90,91] An increasing experience of IL-1
blockade has been matured over recent times and, after showing its
dramatic efficacy in cryopyrin-associated periodic syndrome, a rare and
totally IL-1-mediated hereditary autoinflammatory disorder,[92]
several case reports and case series dedicated to FMF have documented
both efficacy and safety of anti-IL-1 agents, such as anakinra,
canakinumab and rilonacept in patients inadequately responding to
colchicine.[93-96] All IL-1 inhibitors are effective
for controlling attacks and inflammatory activity in patients with
refractory FMF and even in those complicated with AA amyloidosis,
reducing or stabilizing the amount of proteinuria and preserving renal
function in short-term follow-up studies. Structured scores rating FMF
activity or severity by the use of attack parameters (site, duration
and frequency) have been used to classify and settle disease diversity,
while response to treatment has been evaluated by
patients’/parents'/physicians' global assessment of disease severity,
laboratory parameters performed every 6 months, and different scores
related to symptoms and organ damages.[97-102]
How to Put Patients in Differential Diagnosis
Diagnosis
of FMF is mainly a clinical process based on the fever recurrence
combined with different symptoms: the final phenotype is not determined
by the MEFV genotype alone,
but a combination with other modifier genes and environmental factors.
In addition, many diseases share recurrent fever as a common presenting
feature. The problem remains relevant at whatever age, but mostly in
children who frequently may show recurrent infections requiring
assessment and even hospitalization.[103] A frequent
and misdiagnosed cause of recurrent fevers in the pediatric age is
PFAPA syndrome, but a detailed history-taking combined with a mindful
collection of physical findings during flares is crucial to identify
FMF. It is important to be aware that colchicine prophylaxis has been
adopted by different research groups on PFAPA patients, although
complete responses have not been observed.[104] The
discrimination between PFAPA syndrome during childhood, early
onset-Behçet’s disease, and FMF may be challenging: in particular,
Behçet’s disease involves primarily the oral and genital mucosa, but
also skin and eyes.[105] An Israelian large-scale
population-based study has shown that FMF is more frequently diagnosed
in female patients with Behçet’s disease of Arab descent.[106]
Quite similar to FMF is mevalonate kinase
deficiency/hyperimmunoglobulin D syndrome, though the median age at the
onset of this disease is 0.5 years, and these children have concurrent
clinical symptoms involving the gastrointestinal, mucocutaneous and
musculoskeletal system lasting more than three days:[107]
increased urinary levels of mevalonate during febrile flares can
consent to identify patients with mevalonate kinase deficiency.[108]
Another autoinflammatory disorder to differentiate is tumor necrosis
factor receptor-associated periodic syndrome: its presenting features
include a high-grade fever of much longer duration (in comparison with
FMF), abdominal pain, arthralgia, myalgia caused by monocytic
fasciitis, conjunctivitis and periorbital edema, though the genetic
diagnosis remains discriminating.[109] The quite
characteristic recurrence of abdominal pain in FMF patients should also
require to consider alternative diagnosis, such as inflammatory bowel
disease, biliary and renal lithiasis, cholecystitis, pancreatitis,
hemolytic syndromes, Behçet’s disease and acute hepatic porphyrias,
which are rare inborn errors of heme biosynthesis characterized by
acute neurovisceral attacks heralded by severe abdominal pain.
Porphyria is commonly diagnosed during acute attacks by the
demonstration of a striking urinary elevation of the neurotoxic
porphyrin precursors aminolevulinic acid and porphobilinogen, that
correlate with severity of abdominal symptoms.[110] General Conclusions
In
our modern era, FMF cannot be considered a rare condition confined to
the Eastern Mediterranean countries as in the more recent past: indeed,
this disease is scattered throughout the world showing a dynamic face
with genetic and environmental aspects which are diversely tangled to
determine its clinical phenotype.[111] Colchicine
prophylaxis allows all age-patients with FMF to live a normal life with
no restriction or substantial risk of sequelae. However, the
undiagnosed disease has a negative influence on patients’ and their
families’ quality of life because of frequent hospitalizations,
potential surgical interventions caused by a wrong interpretation of
FMF clinical signs or simply due to the recurrence of febrile attacks,
which reduce school or work attendance and limit any social attitudes
for patients.[112] Diagnosis of FMF is mainly made
on the basis of the typical clinical findings in association with the
peculiar ethnicity, family history, and response to colchicine.[113,114]
Despite the great steps in our understanding of FMF, we still have a
number of hanging questions: for instance, what is the exact role of
additional genes in the definition of the final FMF phenotype, what is
the pathophysiology of the disease in patients with only one MEFV mutation or in those without any MEFV
mutation, which are further unexplored inflammatory pathways which
might be involved in the disease expression or progression to
amyloidosis, and so on. The progress in the knowledge of genetic
determinants of FMF could constitute a significant step towards the
understanding of the human genome power and general mechanisms of
inflammation with future relevant therapeutic implications. Wider
awareness of FMF will probably reduce the diagnostic delay in
recognition of the disease and positively affect the quality of life of
patients who will have a lower risk of long-term morbidity and
complications. References
- Stojanov S, Kastner DL. Familial autoinflammatory
diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol
2005;17:586-99. https://doi.org/10.1097/bor.0000174210.78449.6b PMid:16093838
- Ozen S, Demir S. Monogenic periodic fever syndromes: treatment options for the pediatric patient. Paediatr Drugs 2017;19:303-1. https://doi.org/10.1007/s40272-017-0232-6
- Rigante
D. The broad-ranging panorama of systemic autoinflammatory disorders
with specific focus on acute painful symptoms and hematologic
manifestations in children. Mediterr J Hematol Infect Dis 2018;10 https://doi.org/10.4084/mjhid.2018.067
- Rigante D. A developing portrait of hereditary periodic fevers in childhood. Expert Opin Orphan Drugs 2018;6:47-55. https://doi.org/10.1080/21678707.2018.1406797
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949;12:234-47. PMid:18124924
- Reimann
HA, Moadie J, Semerdjian S, Sahyoun PF. Periodic peritonitis; heredity
and pathology: report of seventy-two cases. J Am Med Assoc
1954;154:1254-9. https://doi.org/10.1001/jama.1954.02940490018005 PMid:13151833
- Heller H, Sohar E, Sherf L. Familial Mediterranean fever. AMA Arch Int Med 1958;102:50-71. https://doi.org/10.1001/archinte.1958.00260190052007 PMid:13558745
- Babior BM, Matzner Y. The familial Mediterranean fever gene - cloned at last. N Engl J Med 1997;337:1548-9. https://doi.org/10.1056/NEJM199711203372112 PMid:9366590
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 2017;17:25-31. https://doi.org/10.1038/ng0997-25 PMid:9288094
- The
International FMF Consortium. Ancient missense mutations in a new
member of the RoRet gene family are likely to cause familial
Mediterranean fever. Cell 1997;90:797-807. https://doi.org/10.1016/S0092-8674(00)80539-5 PMid:9288758
- Booth DR, Gillmore JD, Booth SE, et al. Pyrin/marenostrin mutations in familial Mediterranean fever. QJM 1998;91:603-6. https://doi.org/10.1093/qjmed/91.9.603 PMid:10024914
- Rigante D. The fresco of autoinflammatory diseases from the pediatric perspective. Autoimmun Rev 2012;11:348-56. https://doi.org/10.1016/j.autrev.2011.10.008 PMid:22024500
- Rigante
D. The protean visage of systemic autoinflammatory syndromes: a
challenge for inter-professional collaboration. Eur Rev Med Pharmacol
Sci 2010;14:1-18. PMid:20184084
- Fujikura
K. Global epidemiology of familial Mediterranean fever mutations using
population exome sequences. Mol Genet Genomic Med 2015;3:272-82. https://doi.org/10.1002/mgg3.140 PMid: PMC4521964
- Touitou I. The spectrum of familial Mediterranean fever mutations. Eur J Hum Genet 2001;9:473-83. https://doi.org/10.1038/sj.ejhg.5200658n PMid:11464238
- Gershoni-Baruch
R, Shinawi M, Leah K, et al. Familial Mediterranean fever: prevalence,
penetrance and genetic drift. Eur J Hum Genet 2001;9:634-7. https://doi.org/10.1038/sj.ejhg.5200672 PMid:11528510
- Lidar
M, Livneh A. Familial Mediterranean fever: clinical, molecular and
management advancements. Neth J Med 2007;65:318-24. PMid:17954950
- Manna
R, Cerquaglia C, Curigliano V, et al. Clinical features of familial
Mediterranean fever: an Italian overview. Eur Rev Med Pharmacol Sci
2009;13 Suppl 1:51-3. PMid:19530512
- Rigante
D, Frediani B, Galeazzi M, et al. From the Mediterranean to the sea of
Japan: the transcontinental odyssey of autoinflammatory diseases.
Biomed Res Int 2013;2013:485103. https://doi.org/10.1155/2013/485103 PMid:23971037
- Balci-Peynircioglu
B, Akkaya-Ulum YZ, Avci E, et al. Potential role of pyrin, the protein
mutated in familial Mediterranean fever, during inflammatory cell
migration. Clin Exp Rheumatol 2018;36 (6 Suppl 115):116-24.
PMid:30582517
- Tunca M, Akar S, Onen F,
et al. Familial Mediterranean fever in Turkey: results of a nationwide
multicenter study. Medicine 2005 84:1-11. https://doi.org/10.1097/01.md.0000152370.84628.0c PMid:15643295
- Lucherini
OM, Rigante D, Sota J, et al. Updated overview of molecular pathways
involved in the most common monogenic autoinflammatory diseases. Clin
Exp Rheumatol 2018;36 Suppl 110(1):3-9. PMid: 29742053
- Alghamdi M. Familial Mediterranean fever, review of the literature. Clin Rheumatol 2017;36:1707-13. https://doi.org/10.1007/s10067-017-3715-5 PMid: 28624931
- Bagci
S, Toy B, Tuzun A, et al. Continuity of cytokine activation in patients
with familial Mediterranean fever. Clin Rheumatol 2004;23:333-7. https://doi.org/10.1007/s10067-004-0925-4 PMid:15293095
- Kelesoglu
FM, Aygun E, Okumus NK, et al. Evaluation of subclinical inflammation
in familial Mediterranean fever patients: relations with mutation types
and attack status: a retrospective study. Clin Rheumatol
2016;35:2757-63. https://doi.org/10.1007/s10067-016-3275-0 PMid:27106545
- Van
Gorp H, Saavedra PH, de Vasconcelos NM, et al. Familial Mediterranean
fever mutations lift the obligatory requirement for microtubules in
pyrin inflammasome activation. Proc Natl Acad Sci USA 2016;113:14384-9.
https://doi.org/10.1073/pnas.1613156113 PMid:27911804
- Park
YH, Wood G, Kastner DL, et al. Pyrin inflammasome activation and RhoA
signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol
2016;17:914-21. https://doi.org/10.1038/ni.3457 PMid:27270401
- Demirtürk
L, Ozel AM, Cekem K, et al. Co-existence of Helicobacter pylori
infection in patients with familial Mediterranean fever (FMF) and the
effect of Helicobacter pylori on the frequency and severity of FMF
attacks. Dig Liver Dis 2005;37:153-8. https://doi.org/10.1016/j.dld.2004.09.027 PMid:15888278
- Verrecchia
E, Sicignano LL, La Regina M, et al. Small intestinal bacterial
overgrowth affects the responsiveness to colchicine in familial
Mediterranean fever. Mediators Inflamm 2017;2017:7461426. https://doi.org/10.1155/2017/7461426 PMid:29379228
- Rigante
D, La Torraca I, Avallone L, et al. The pharmacological basis of
treatment with colchicine in children with familial Mediterranean
fever. Eur Rev Med Pharmacol Sci 2006;10:173-8. PMid:16910346
- Rigante
D. A systematic approach to autoinflammatory syndromes: a spelling
booklet for the beginner. Expert Rev Clin Immunol 2017;13:571-97. https://doi.org/10.1080/1744666X.2017.1280396 PMid:28064547
- Dodé
C, Pecheux C, Cazeneuve C, et al. Mutations in the MEFV gene in a large
series of patients with a clinical diagnosis of familial Mediterranean
fever. Am J Med Genet 2000;92:241-6. PMid: 10842288 https://doi.org/10.1002/(SICI)1096-8628(20000605)92:4<241::AID-AJMG3>3.0.CO;2-G
- Shinar
Y, Livneh A, Langevitz P, et al. Genotype-phenotype assessment of
common genotypes among patients with familial Mediterranean fever. J
Rheumatol 2000; 27:1703-7. PMid:10914855
- Pasa
S, Altintas A, Devecioglu B, et al. Familial Mediterranean fever gene
mutations in the Southeastern region of Turkey and their phenotypical
features. Amyloid 2008;15:49-53. https://doi.org/10.1080/13506120701815456 PMid:18266121
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009;61:1447-53. https://doi.org/10.1002/art.24458 PMid:19790133
- Marek-Yagel
D, Bar-Joseph I, Pras E, et al. Is E148Q a benign polymorphism or a
disease-causing mutation? J Rheumatol 2009;36:2372.https://doi.org/10.3899/jrheum.090250 PMid:19820229
- Comak
E, Akman S, Koyun M, et al. Clinical evaluation of R202Q alteration of
MEFV genes in Turkish children. Clin Rheumatol 2014;33:1765-71. https://doi.org/10.1007/s10067-014-2602-6 PMid:24718488
- Gershoni-Baruch
R, Brik R, Zacks N, et al. The contribution of genotypes at the MEFV
and SAA1 loci to amyloidosis and disease severity in patients with
familial Mediterranean fever. Arthritis Rheum 2003;48:1149-55. https://doi.org/10.1002/art.10944 PMid:12687559
- Caso
F, Costa L, Rigante D, et al. Caveats and truths in genetic, clinical,
autoimmune and autoinflammatory issues in Blau syndrome and early onset
sarcoidosis. Autoimmun Rev 2014;13:1220-9. https://doi.org/10.1016/j.autrev.2014.08.010 PMid:25182201
- Berkun
Y, Karban A, Padeh S, et al. NOD2/CARD15 gene mutations in patients
with familial Mediterranean fever. Semin Arthritis Rheum 2012;42:84-8. https://doi.org/10.1016/j.semarthrit.2011.12.002 PMid:22244368
- Rigante D. New mosaic tiles in childhood hereditary autoinflammatory disorders. Immunol Lett 2018;193:67-76. https://doi.org/10.1016/j.imlet.2017.11.013 PMid:29198619
- Rigante
D, La Torraca I, Ansuini V, et al. The multi-face expression of
familial Mediterranean fever in the child. Eur Rev Med Pharmacol Sci
200;10:163-71. PMid:16910345
- Soriano A, Manna R. Familial Mediterranean fever: new phenotypes. Autoimmun Rev 2012;12:31-7. https://doi.org/10.1016/j.autrev.2012.07.019 PMid:22878273
- El-Shanti H, Majeed HA, El-Khateeb M. Familial mediterranean fever in Arabs. Lancet 2006 Mar 25;367(9515):1016-24. https://doi.org/10.1016/S0140-6736(06)68430-4 PMid:16564365
- Adrovic
A, Sahin S, Barut K, et al. Familial Mediterranean fever and periodic
fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome:
shared features and main differences. Rheumatol Int 2019;39:29-36. https://doi.org/10.1007/s00296-018-4105-2 PMid:30019226
- Gentileschi
S, Vitale A, Frediani B, et al. Challenges and new horizons in the
periodic fever, aphthous stomatitis, pharingitis and adenitis (PFAPA)
syndrome. Expert Opin Orphan Drugs 2017;5:165-71. https://doi.org/10.1080/21678707.2017.1279049
- Gaggiano
C, Rigante D, Sota J, et al. Treatment options for periodic fever,
aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA)
syndrome in children and adults: a narrative review. Clin Rheumatol
2019; 38: 11-17. https://doi.org/10.1007/s10067-018-4361-2 PMid:30488366
- Cantarini
L, Vitale A, Bartolomei B, et al. Diagnosis of PFAPA syndrome applied
to a cohort of 17 adults with unexplained recurrent fevers. Clin Exp
Rheumatol 2012;30:269-71. PMid:22325152
- Rigante
D, Vitale A, Natale MF, et al. A comprehensive comparison between
pediatric and adult patients with periodic fever, aphthous stomatitis,
pharyngitis, and cervical adenopathy (PFAPA) syndrome. Clin Rheumatol
2017;36:463-8. https://doi.org/10.1007/s10067-016-3317-7 PMid:27251674
- Cakir
M, Ozgenc F, Baran M, et al. A rare cause of refractory ascites in a
child: familial Mediterranean fever. Rheumatol Int 2010;30:531-4. https://doi.org/10.1007/s00296-009-0957-9 PMid:19466424
- Reissman
P, Durst Al, Rivkind A, et al. Elective laparoscopic appendectomy in
patients with familial Mediterranean fever. World J Surg
1994;18:139-42. https://doi.org/10.1007/BF00348205 PMid:8197770 PMid:8197770
- Kees
S, Langevitz P, Zemer D, et al. Attacks of pericarditis as a
manifestation of familial Mediterranean fever. QJM 1997;90:643-7. PMid:
9415347 https://doi.org/10.1093/qjmed/90.10.643 PMid:9415347
- Rigante D, Cantarini L, Imazio M, et al. Autoinflammatory diseases and cardiovascular manifestations. Ann Med 2011;43:341-6. https://doi.org/10.3109/07853890.2010.547212 PMid:21284530
- Eshel
G, Vinograd I, Barr J, et al. Acute scrotal pain complicating familial
Mediterranean fever in children. Br J Surg 1994; 81: 894-6. https://doi.org/10.1002/bjs.1800810633 PMid:8044614
- Jarjour
RA, Dodaki R. Arthritis patterns in familial Mediterranean fever
patients and association with M694V mutation. Mol Biol Rep
2011;38:2033-6. https://doi.org/10.1007/s11033-010-0326-5 PMid:20845072
- Rigante D, Cantarini L. Monogenic autoinflammatory syndromes at a dermatological level. Arch Dermatol Res 2011;303:375-80. https://doi.org/10.1007/s00403-011-1134-z PMid:21340744
- Kotevoglu
N, Sahin F, Ozkiris SO, et al. Protracted febrile myalgia of familial
Mediterranean fever. Clin Exp Rheumatol 2004;22:S69-S70. PMid:15515790
- Tufan G, Demir S. Uncommon clinical pattern of FMF: protracted febrile myalgia syndrome. Rheumatol Int 2010;30:1089-90. https://doi.org/10.1007/s00296-009-1024-2 PMid:19590876
- Rigante
D, Lopalco G, Tarantino G, et al. Non-canonical manifestations of
familial Mediterranean fever: a changing paradigm. Clin Rheumatol
2015;34:1503-11. https://doi.org/10.1007/s10067-015-2916-z PMid:25761640
- Feld
O, Yahalom G, Livneh A. Neurologic and other systemic manifestations in
FMF: published and own experience. Best Pract Res Clin Rheumatol
2012;26:119-33. https://doi.org/10.1016/j.berh.2012.01.004 PMid:22424198
- Medlej-Hashim
M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean
fever patients: correlation with MEFV genotype and SAA1 and MICA
polymorphisms effects. BMC Med Genet 2004;5:4. https://doi.org/10.1186/1471-2350-5-4 PMid:15018633 PMCid:PMC356915
- Touitou
I, Sarkisian T, Medlej-Hashim M, et al. Country as the primary risk
factor for renal amyloidosis in familial Mediterranean fever. Arthritis
Rheum 2007;56:1706-12. https://doi.org/10.1002/art.22507 PMid:17469185
- Rigante
D, Frediani B, Cantarini L. A comprehensive overview of the hereditary
periodic fever syndromes. Clin Rev Allergy Immunol 2018;54:446-53. https://doi.org/10.1007/s12016-016-8537-8 PMid:27068928
- Lange-Sperandio
B, Möhring K, Gutzler F, et al. Variable expression of vasculitis in
siblings with familial Mediterranean fever. Pediatr Nephrol
2004;19:539-43. https://doi.org/10.1007/s00467-004-1440-1 PMid:15015067
- Alghamdi
M. Autoinflammatory disease-associated vasculitis/vasculopathy. Curr
Rheumatol Rep 2018;20:87. doi: 10.1007/s11926-018-0788-3 https://doi.org/10.1007/s11926-018-0788-3 PMid:30446874
- Yazici
A, Cefle A, Savli H. The frequency of MEFV gene mutations in Behçet's
disease and their relation with clinical findings. Rheumatol Int
2012;32:3025-30. https://doi.org/10.1007/s00296-011-2011-y PMid:21901355
- Soriano A, Pras E. Familial Mediterranean fever: genetic update. Isr Med Assoc J 2014;16:274-6. PMid: 24979829
- Stabile
A, Bertoni B, Ansuini V, et al. The clinical spectrum and treatment
options of macrophage activation syndrome in the pediatric age. Eur Rev
Med Pharmacol Sci 2006;10:53-9. PMid:16705949
- Rigante
D, Emmi G, Fastiggi M, et al. Macrophage activation syndrome in the
course of monogenic autoinflammatory disorders. Clin Rheumatol
2015;34:1333-9. https://doi.org/10.1007/s10067-015-2923-0 PMid:25846831
- Brenner
R, Ben-Zvi I, Shinar Y, et al. Familial Mediterranean fever and
incidence of cancer: an analysis of 8,534 Israeli patients with 258,803
person-years. Arthritis Rheumatol 2018;70:127-33. https://doi.org/10.1002/art.40344 PMid:28992365
- Sohar
E, Gafni J, Pras M, et al. Familial Mediterranean fever. A survey of
470 cases and review of the literature. Am J Med 1967;43:227-53. https://doi.org/10.1016/0002-9343(67)90167-2 PMid:5340644
- Livneh
A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial
Mediterranean fever. Arthritis Rheum 1997;40:1879-85. https://doi.org/10.1002/1529-0131(199710)40 PMid:9336425
- Yalçinkaya
F, Ozen S, Ozçakar ZB, et al. A new set of criteria for the diagnosis
of familial Mediterranean fever in childhood. Rheumatology (Oxford)
2009;48:395-8. https://doi.org/10.1093/rheumatology/ken509 PMid:19193696
- Demirkaya
E, Saglam C, Turker T, et al. Performance of different diagnostic
criteria for Familial Mediterranean fever in children with periodic
fevers: results from a multicenter international registry. J Rheumatol
2016;43:154-60. https://doi.org/10.3899/jrheum.141249 PMid:26568587
- Federici
S, Sormani MP, Ozen S, et al. Evidence-based provisional clinical
classification criteria for autoinflammatory periodic fevers. Ann Rheum
Dis 2015;74:799-805. https://doi.org/10.1136/annrheumdis-2014-206580 PMid:25637003
- Yepiskoposyan L, Harutyunyan A. Population genetics of familial Mediterranean fever: a review. Eur J Hum Genet 2007; 15: 911-6. https://doi.org/10.1038/sj.ejhg.5201869 PMid:17568393
- Caso
F, Cantarini L, Lucherini OM, et al. Working the endless puzzle of
hereditary autoinflammatory disorders. Mod Rheumatol 2014;24:381-9. https://doi.org/10.3109/14397595.2013.843755 PMid:24251993
- Hentgen
V, Grateau G, Stankovic-Stojanovic K, et al. Familial Mediterranean
fever in heterozygotes: are we able to accurately diagnose the disease
in very young children? Arthritis Rheum 2013;65:1654-62. https://doi.org/10.1002/art.37935 PMid:23508419
- Zemer
D, Pras M, Sohar E, et al. Colchicine in the prevention and treatment
of the amyloidosis of familial Mediterranean fever. N Engl J Med
1986;314:1001-5. https://doi.org/10.1056/NEJM198604173141601 PMid:3515182
- Rigante D, Vitale A, Lucherini OM, et al. The hereditary autoinflammatory disorders uncovered. Autoimmun Rev 2014;13:892-900. https://doi.org/10.1016/j.autrev.2014.08.001 PMid:25149390
- Knieper
AM, Klotsche J, Lainka E, et al. Familial Mediterranean fever in
children and adolescents: factors for colchicine dosage and predicting
parameters for dose increase. Rheumatology (Oxford) 2017;56:1597-606. https://doi.org/10.1093/rheumatology/kex222 PMid:2885939
- Yanmaz MN, Özcan AJ, Savan K. The impact of familial Mediterranean fever on reproductive system. Clin Rheumatol 2014;33:1385-8. https://doi.org/10.1007/s10067-014-2709-9 PMid:24924605
- Ozturk
MA, Kanbay M, Kasapoglu B, et al. Therapeutic approach to familial
Mediterranean fever: a review update. Clin Exp Rheumatol 2011;29(4
Suppl 67):S77-86. PMid:21968242
- Eroglu
FK, Beşbaş N, Topaloglu R, et al. Treatment of colchicine-resistant
Familial Mediterranean fever in children and adolescents. Rheumatol Int
2015;35:1733-7. https://doi.org/10.1007/s00296-015-3293-2 PMid:26001859
- Corsia
A, Georgin-Lavialle S, Hentgen V, et al. A survey of resistance to
colchicine treatment for French patients with familial Mediterranean
fever. Orphanet J Rare Dis 2017;12:54. https://doi.org/10.1186/s13023-017-0609-1 PMid:28302131
- Cerquaglia
C, Diaco M, Nucera G, et al. Pharmacological and clinical basis of
treatment of familial Mediterranean fever (FMF) with colchicine or
analogues: an update. Curr Drug Targets Inflamm Allergy 2005;4:117-24. https://doi.org/10.2174/1568010053622984 PMid:15720245 PMid:15720245
- Ben-Zvi
I, Krichely-Vachdi T, Feld O, et al. Colchicine-free remission in
familial Mediterranean fever: featuring a unique subset of the
disease-a case control study. Orphanet J Rare Dis 2014;9:3. doi:
10.1186/1750-1172-9-3 https://doi.org/10.1186/1750-1172-9-3 PMid:24401676
- Tamir
N, Langevitz P, Zemer D, et al. Late-onset familial Mediterranean fever
(FMF): a subset with distinct clinical, demographic, and molecular
genetic characteristics. Am J Med Genet 1999;87:30-5. PMid: 10528243 https://doi.org/10.1002/(SICI)1096-8628(19991105)87:1<30::AID-AJMG6>3.0.CO;2-B
- Rigante
D, Manna R. A position for tumor necrosis factor inhibitors in the
management of colchicine-resistant familial Mediterranean fever?
Immunol Lett 2016;180:77-8. https://doi.org/10.1016/j.imlet.2016.10.007 PMid:27984066
- Lopalco
G, Cantarini L, Vitale A, et al. Interleukin-1 as a common denominator
from autoinflammatory to autoimmune disorders: premises, perils, and
perspectives. Mediators Inflamm 2015;2015:194864. https://doi.org/10.1155/2015/194864 PMid:25784780
- Cantarini
L, Lopalco G, Cattalini M, et al. Interleukin-1: Ariadne's thread in
autoinflammatory and autoimmune disorders. Isr Med Assoc J
2015;17:93-7. PMid:26223084
- Cantarini L,
Lucherini OM, Frediani B, et al. Bridging the gap between the clinician
and the patient with cryopyrin-associated periodic syndromes. Int J
Immunopathol Pharmacol 2011;24:827-36. https://doi.org/10.1177/039463201102400402 PMid:22230390
- Varan
Ö, Kucuk H, Babaoglu H, et al. Efficacy and safety of interleukin-1
inhibitors in familial Mediterranean fever patients complicated with
amyloidosis. Mod Rheumatol 2018 Apr 27:1-4. https://doi.org/10.1080/14397595.2018.1457469 PMid:29578360
- De
Benedetti F, Gattorno M, Anton J, et al. Canakinumab for the treatment
of autoinflammatory recurrent fever syndromes. N Engl J Med
2018;378:1908-19. https://doi.org/10.1056/NEJMoa1706314 PMid:29768139
- Vitale
A, Insalaco A, Sfriso P, et al. A snapshot on the on-label and
off-label use of the interleukin-1 inhibitors in Italy among
rheumatologists and pediatric rheumatologists: a nationwide
multi-center retrospective observational study. Front Pharmacol
2016;7:380. https://doi.org/10.3389/fphar.2016.00380 PMid:27822185
- Hashkes
PJ, Spalding SJ, Hajj-Ali R, et al. The effect of rilonacept versus
placebo on health-related quality of life in patients with poorly
controlled familial Mediterranean fever. Biomed Res Int
2014;2014:854842. https://doi.org/10.1155/2014/854842, PMid:25147819
- Mor
A, Shinar Y, Zaks N, et al. Evaluation of disease severity in familial
Mediterranean fever. Semin Arthritis Rheum 2005;35:57-64. https://doi.org/10.1016/j.semarthrit.2005.02.002 PMid:16084225
- Cantarini
L, Lucherini OM, Iacoponi F, et al. Development and preliminary
validation of a diagnostic score for identifying patients affected with
adult-onset autoinflammatory disorders. Int J Immunopathol Pharmacol
2010;23:1133-4. https://doi.org/10.1177/039463201002300417 PMid:21244762
- Cantarini
L, Iacoponi F, Lucherini OM, et al. Validation of a diagnostic score
for the diagnosis of autoinflammatory diseases in adults. Int J
Immunopathol Pharmacol 2011;24:695-702. https://doi.org/10.1177/039463201102400315 PMid:21978701
- Piram
M, Koné Paut I, Lachmann H, et al. Validation of the auto-inflammatory
diseases activity index (AIDAI) for hereditary recurrent fever
syndromes. Ann Rheum Dis 2014;73:2168-73. https://doi.org/10.1136/annrheumdis-2013-203666 PMid:24026675
- Ter
Haar NM, Annink KV, Al-Mayouf SM, et al. Development of the
autoinflammatory disease damage index (ADDI). Ann Rheum Dis
2017;76:821-30. https://doi.org/10.1136/annrheumdis-2016-210092 PMid:27811147
- Ter
Haar NM, van Delft ALJ, Annink KV, et al. In silico validation of the
Autoinflammatory Disease Damage Index. Ann Rheum Dis 2018;77:1599-1605.
https://doi.org/10.1136/annrheumdis-2018-213725 PMid:30077992
- Rigante
D. Autoinflammatory syndromes behind the scenes of recurrent fevers in
children. Med Sci Monit 2009;15:RA179-87. PMid:19644432
- Dusser
P, Hentgen V, Neven B, et al. Is colchicine an effective treatment in
periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis
(PFAPA) syndrome? Joint Bone Spine 2016;83:406-11. https://doi.org/10.1016/j.jbspin.2015.08.017 PMid:27068612
- Caso
F, Costa L, Rigante D, et al. Biological treatments in Behçet's
disease: beyond anti-TNF-therapy. Mediators Inflamm 2014;2014:107421. https://doi.org/10.1155/2014/107421 PMid:25061259
- Watad
A, Tiosano S, Yahav D, et al. Behçet's disease and familial
Mediterranean fever: Two sides of the same coin or just an association?
A cross-sectional study. Eur J Intern Med 2017;39:75-8. https://doi.org/10.1016/j.ejim.2016.10.011 PMid:27776949
- Ter
Haar NM, Jeyaratnam J, Lachmann HJ, et al. The phenotype and genotype
of mevalonate kinase deficiency: a series of 114 cases from the
Eurofever Registry. Arthritis Rheumatol 2016;68:2795-805. https://doi.org/10.1002/art.39763 PMid:27213830
- Esposito
S, Ascolese B, Senatore L, et al. Current advances in the understanding
and treatment of mevalonate kinase deficiency. Int J Immunopathol
Pharmacol 2014;27:491-8. https://doi.org/10.1177/039463201402700404 PMid:25572728
- Rigante
D, Lopalco G, Vitale A, et al. Key facts and hot spots on tumor
necrosis factor receptor-associated periodic syndrome. Clin Rheumatol
2014;33:1197-207. https://doi.org/10.1007/s10067-014-2722-z PMid:24935411
- Naik
H, Stoecker M, Sanderson SC, et al. Experiences and concerns of
patients with recurrent attacks of acute hepatic porphyria: A
qualitative study. Mol Genet Metab 2016;119:278-83. https://doi.org/10.1016/j.ymgme.2016.08.006 PMid:27595545
- Cantarini
L, Rigante D, Brizi MG, et al. Clinical and biochemical landmarks in
systemic autoinflammatory diseases. Ann Med 2012;44:664-73. https://doi.org/10.3109/07853890.2011.598546 PMid:21972825
- Cantarini
L, Vitale A, Lucherini OM, et al. Childhood versus adulthood-onset
autoinflammatory disorders: myths and truths intertwined. Reumatismo
2013;65:55-62. https://doi.org/10.4081/reumatismo.2013.55 PMid:23877409
- Rigante
D, Vitale A, Natale MF, et al. Lights and shadows in autoinflammatory
syndromes from the childhood and adulthood perspective. Clin Rheumatol
2016;35:565-72. https://doi.org/10.1007/s10067-015-3132-6 PMid: 6631101
- Vitale
A, Rigante D, Lucherini OM, et al. The diagnostic evaluation of
patients with a suspected hereditary periodic fever syndrome:
experience from a referral center in Italy. Intern Emerg Med
2017;12:605-11. https://doi.org/10.1007/s11739-017-1622-z PMid:28194697
[TOP]