Federica Pilo1 and Emanuele Angelucci2.
1 Hematology and Transplant Center, Ospedale Oncologico “Armando Businco” Cagliari, Italy,
2 Hematology and Transplant Center, IRCCS Ospedale Policlinico San Martino, Genova, Italy.
Correspondence to: Federica Pilo. Hematology and Transplant Center, Ospedale Oncologico “Armando Businco” Cagliari, Italy,
Published: May 1, 2019
Received: February 5, 2019
Accepted: March 21, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019030 DOI
10.4084/MJHID.2019.030
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
issue of iron overload in hemopoietic cell transplantation has been
first discussed in the field of transplantation for thalassemia.
Thalassemia major is characterized by ineffective erythropoiesis and
hemolysis leading to severe anemia. Patients require regular blood
transfusion therefore they develop iron overload causing organ damage
and hematopoietic cell transplantation (HCT) is a consolidated
reliably curative option.
In this category of patients an
important issue for transplant outcome is the iron burden before
transplant and in the long-life post-transplant. Nevertheless today the
concept of the impact of iron overload / toxicity on the outcome of HCT
has been extended to other diseases characterized by periods of
variable duration of transfusion dependence .
Recent preclinical
data has shown how increased production of reactive oxygen species
(ROS) resulting under iron overload condition, could impair the stem
cells clonality capacity, proliferation and maturation. Also,
microenvironment cells could be affected through this mechanism. For
this reason, iron overload is becoming an important issue also in the
engraftment period post-transplant.
The aim of this review is to
update consolidated knowledge about the role of iron overload/iron
toxicity in the HCT setting in non-malignant and in malignant
diseases introducing the concept of exposition of free toxic iron forms
and related cellular damage in the different stage of transplant.
|
Introduction
ß-thalassemias
are a group of genetic hemoglobinopathy presenting different
grading of ineffective erythropoiesis and hemolysis leading to anemia
in the majority of patients. ß-Thalassemia Major (ß-TM) is the most
common form of thalassemia. Patients affected by ß-TM require regular
blood transfusion therefore they develop iron overload causing organ
damage.[1] This condition is therefore defined as Transfusion Dependent Thalassemia (TDT).
More
than 30 years have passed since the first successful Hematopoietic Cell
Transplantation (HCT) in thalassemia. Since then, more than 3000
transplants have been performed worldwide with outstanding results.[2] Gene therapy has been now finally developed and promises to be a further improvement in thalassemia cure option.[3]
Firstly
in this category of patients iron overload toxicity has emerged as an
important issue for transplant outcome. Clinical results revealed how
iron exceeded before HCT affects the outcome in TDT. Subsequently the
role of iron overload / toxicity have been investigated in others
transfusion dependent diseases including myelodysplastic syndromes
(MDS). It is well known that iron overload is deleterious for organs
such as liver, heart and endocrine glands and, it has been postulated
could also increases the risk of infections and Graft versus Host
Disease (GvHD) early after HCT. High baseline Ferritin levels before
HCT have been shown to negatively influence clinical outcome in
diseases different from TDT.[4-6]
If solid
clinical evidence has established the negative impact of high iron
burden and related tissue damage on the outcome of HCT for TDT, recent
preclinical data has shown how increased production of reactive oxygen
species (ROS) resulting under iron overload condition, could impair the
hemopoietic niche and therefore the stem cells clonality capacity,
proliferation and maturation independently from baseline disease. Also,
microenvironment cells could be affected through this mechanism. Moving
from this hypothesis, in-vitro and animal model experimental studies
started to understand if iron toxicity could be an issue also in the
HCT engraftment period.[7]
The aim of this review
is to update consolidated knowledge about the role of iron
overload/iron toxicity in the HCT setting introducing the concept of
exposition of free toxic iron forms and related cellular damage.
Recently Understood Mechanisms Support Iron Toxicity and Cell Damage
Iron
is an essential element for the normal cellular life but when it
exceeds the needs of physiological cellular processes, reactive oxygen
species are produced. Increased ROS levels may have beneficial or toxic
effects depending on their levels. In the case of imbalance in
intracellular redox homeostasis, ROS levels overwhelm cellular
antioxidant defenses, and an oxidative stress state is established.
Numerous cellular functions are determined upon appropriate
intracellular ROS levels, and are deregulated under oxidative stress
conditions. These processes involve the activation of signaling
pathways leading to alterations of cellular cycle, proliferation,
differentiation, and eventually cell death.[7]
Iron in Phisiological and Pathological Conditions
Thanks
to its biochemical feature, iron moves across different oxidation
states (ranging from Fe2- to Fe6+; the two common forms involved in
human biochemical reaction are Fe2+ and Fe3+), this flexibility makes
iron suitable for a variety of normal biochemical reactions
particularly involving electron transport and mitochondrion activities.
To maintain a stable level of iron and a normal iron homeostasis,
living organisms must be able to release stored iron during iron
deficiency and store excess iron during iron sufficiency in an
appropriate manner. Under normal condition iron circulates binding to
transferrin and systemic iron balance is mainly maintained by the iron
regulatory hormone hepcidin that binding to ferroportin (FPN) on the
cell surface, regulates the iron cellular efflux. When iron overwhelms
the transferrin’s capacity to transport iron, non-transferrin bound
iron (NTBI) appears in the circulation and together with its active
biological component, labile plasma iron (LPI), is able to enter the
cell through canonical routes but also through alternative channels
participating to increase the labile cell iron (LCI) pool (Table 1). LCI is the intracellular free iron form that contributes to the mitochondrial life, leading to hemoglobin production,[8]
energy production throughout the Krebs cycle and iron sulfuric group
formation that are fundamental for the DNA synthesis and
duplication.
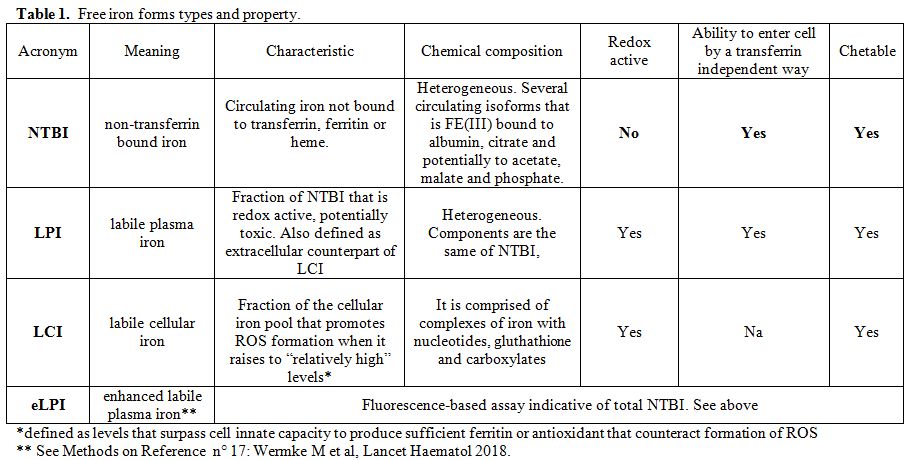 |
Table
1. Free iron forms types and property. |
The
exceed iron, is deposited in the stable ferritin form. Excess LCI
can enter the mitochondria and take part to increase the cellular ROS
production through the Fenton’s reaction. Usually the cell holds
physiological adaptation mechanisms against the ROS level increases,
mainly mediated by the activation of nuclear factor erythroid 2-related
factor (Nrf2). Oxidative stress, normally produced in the cell,
activates the Nrf2 pathway that is able to enter the nucleus and
up-regulate the expression of antioxidant enzymes. Although Nrf2 can
protect normal cells from oxidative stress, when the LCI level
increases this mechanism becomes inadequate to control the ROS surplus
and an oxidative stress state is established in the cell. Maintaining
ROS at an appropriate level plays an important role in biological
phenomena; increasing ROS or damage of the antioxidant system could
lead to oxidative stress reactions and finally to cell damage (Figure 1).[9]
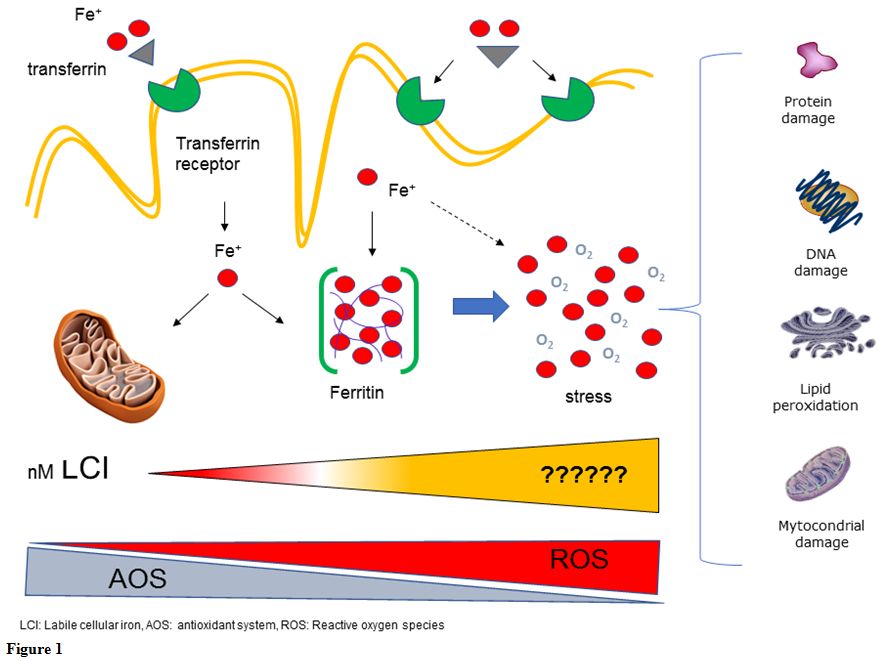 |
Figure
1. |
Nowadays,
ferritin is considered a steady and not biological active form of iron,
while LPI is considered the main trigger of cell damage more
representative of the dynamic tissue damage. The scientific community
is moving the iron disease from a “Bulky” disease, such as
classically in thalassemia (based on quantitative iron parameters as
ferritin, red blood cell transfusion number, MRI) to a “toxic” disease
(based on active and dynamic biological markers as NTBI/LPI). The mechanisms leading to iron dependent tissue / cells toxicity have been recently summarized by the following equation:
Iron toxicity tissue = Σ Tissue reactive iron x Genetics x Environmental Factors x Time
This
formula proposes that tissue toxicity arises from both the quantity of
toxic iron species (i.e., tissue reactive species = NTBI/LPI). The
detrimental effects are further modulated by the host genetic
characteristics, by the individual anti‐oxidant mechanisms and by
environmental factors.[10] Free Iron Toxicity before Transplant
As
far as the clinical counterpart of these biologic phenomena in
transplantation, a clear example is the following: Pesaro’s group
divided TDT patients before HCT in three classes of risk predicting
outcome based on: Liver fibrosis staging, Hepatomegaly presence,
adequate iron chelation. This scoring system received criticism for its
non-quantitative method mainly regarding hepatomegaly and the
definition of adequate chelation.[11,12]
Recently
Angelucci et al revisited the Pesaro TDT score system based on the
above-mentioned new concept of iron toxicity. It is evident that all
the three risk factors were not quantitative direct markers of iron
overload “per se”, but indirect measures of intensity and extent of
tissue exposure to toxic iron. Authors concluded that “adequacy of
chelation” clearly means consistent and sufficient suppression of
tissue reactive iron species (NTBI/LPI) over time. “Liver fibrosis” is
definitely a marker of toxic iron exposure and environmental factors
(i.e., viral infections) in the liver and “Hepatomegaly” reflects the
extent of iron deposition and the time throughout exposure to
NTBI and toxic reactive iron.[12]
At this time
in all the published studies outside TDT, only the correlation between
direct or indirect estimates of iron overload (mainly serum ferritin,
transfusion burden and MRI values) and outcome parameters has been
explored, while the duration of exposure to toxic iron species has not
been taken into account.[4,5,13-16]
The
first study that explored the LPI role in relationship with outcome was
published by Wermke and colleagues in malignancies. They investigated
the predictive value of both stored (MRI-derived liver iron content)
and non-transferrin-bound-iron, defined as enhanced labile plasma iron
(eLPI) (see table 1) on
post-transplantation outcomes in patients with acute myeloid leukemia
(AML) or myelodysplastic syndrome (MDS) . Their prospective,
observational ALLIVE study recruited 112 patients transplanted in three
years and showed that patients who had raised eLPI concentration
at baseline, also had significantly increased incidence of non-relapse
mortality at day 100 (33%) compared with those who had normal eLPI at
baseline (7%) (P= 0.00034).They concluded that peri-transplantation
eLPI-scavenging strategies could be explored in prospective
international clinical trials for patients with systemic iron overload.[17]
Free Iron Toxicity During the Engraftment Period
It
is supposed that LPI may be involved either in the occurrence of
toxicity and other complications commonly observed in the early post
HCT period independently from the underline disease and iron status.[5,18]
Firstly
in patients who underwent HCT for several hematologic
malignancies, it has been demonstrated how LPI levels, although normal
at baseline measurements, increased substantially 48h after the start
of conditioning with a peak around day 0, and remained increased until
engraftment when it returned to baseline levels.[19]
The
fast and substantial increase in LPI levels on Day 0 reflects a
disruption of iron homeostasis by conditioning due to massive iron
release by myeloablation and the temporary lack of iron uptake by the
ablated erythroid system. In addition, it can be speculated that the
conditioning-induced ablation of erythropoiesis could reduce the
synthesis of erythroferrone, an erythroid hormone that suppresses
hepcidin, favoring increased hepcidin levels that block the cellular
iron efflux.[20]
Subsequently, reutilization of
iron by restored erythropoiesis by engraftment leads to a substantial
drop in LPI levels, but not in hepcidin levels, probably modulated also
by inflammation. The interesting information is that ferritin levels
that were already increased at baseline did not change throughout the
engraftment period; so that cytotoxic chemotherapy and subsequent
engraftment in HCT patients leads to changes in LPI but not in
ferritin. For this reason LPI represents the marker that better
reflects the modifications in iron status and could serve as a target
of a possible chelation therapy in the early period of HCT.[21]
Similar results are shown by Duca et al regarding NTBI levels in thalassemic and leukemic patients.[22]
NTBI levels were constantly higher at baseline in the thalassemia group
but the relative increase compared to baseline was higher in leukemia
patients (between 2.2- and 3-fold) than in thalassemia patients
(between 1.6- and 2.5-fold). Early after transplant, concomitant with
erythropoietic recovery, NTBI returned to respective baseline values.
The marked increase of NTBI in serum after myeloablative chemotherapy
was originally attributed to suppression of erythropoietic activity.
Other possible sources of extracellular iron after myeloablative
conditioning include lysis of erythroid bone marrow cells by cytotoxic
injury. Also, this study confirms reduction of the iron uptake by
erythroid precursors during and immediately after chemotherapy.
Furthermore, in this case, NTBI decrement in all the patients after HCT
could be explained by high iron requirement during allogeneic erythroid
marrow rebuilding. High levels of iron overload, in the absence of
chelation, are responsible for the persistence of NTBI high levels even
after marrow reconstitution in thalassemia patients.[22]
A
growing body of evidence describes how iron induced oxidative stress
and increased ROS levels can modulate several signaling pathways (such
as Protein kinase B, Tumor protein p53 and Wnt protein family,) which
promote cell survival, avoid apoptosis, allow escape from growth
arrest, and facilitate cancer transformation. Actually, ROS are
involved in a complicated web of signaling networks where their
generation is regulated by multiple pathways. Conversely,
ROS act as signaling
molecules for other signaling pathways
such as PTEN, PTP1B, MAPK and NF-κB
involved in different ways with the HSCs fate.[23,24]
In
2013, Chai and colleagues established an iron overloaded mouse model to
investigate the effects of iron overload on hematopoietic stem and
progenitor cells (HSPCs). Results show that iron overload markedly
decreased the ratio of immature hematopoietic cells and reduced HSPCs
clonogenic capacity. Iron overload increased ROS levels of HSPCs
through the NOX4/ROS/P38 MAPK signaling pathway.[25]
Similar
results were found using bone marrow mesenchymal cells (BM-MSCs) in a
similar murine model suggesting that iron can impair not only the HSPCs
clonogenic capacity but similarly the quantity and quality of BM-MSCs
and the bone marrow microenvironment as well.[26,27]
The
effect of oxidative stress on hemopoiesis has been investigated in a
murine transplant setting. A murine model was used to investigate the
possible relationship between iron overload and engraftment
post-allogeneic hematopoietic cell transplant. Donor bone marrow
mononuclear cells (BM-MNCs) from iron overloaded mice and normal mice
were transplanted into recipient mice. Flow cytometry analysis of
peripheral blood cells from the recipient mice demonstrated that
recipient mice of iron- overloaded donor had, after transplant, lower
levels of myeloid B and T-lymphocytic lineage engraftments compared to
the recipient mice of normal donor.[25]
A
different conclusion was described by Okabe and colleagues,[26]
who showed in an iron overloaded mouse recipient that oxidative stress
could affect the engraftment of hematopoietic cells from a normal donor
by modifying microenvironment and remarkably reducing expression of
CXCL12, VCAM-1, Kit-ligand, erythropoietin and thrombopoietin. They
concluded that iron overload can damage bone marrow stromal and other
key organs (liver, kidney) and therefore, indirectly, hematopoiesis.
Interestingly,
in almost all the above murine models, hematopoietic insufficiency
improved by treating recipient mice with iron chelator or with the
powerful antioxidant N-acetyl-cysteine (NAC), conveying that iron
overload may be closely related to high oxidative stress.[25]
It
is important to note that these experimental observations have not yet
led to an operative approach. Little literature is available addressing
the issue of peri-transplant chelation in thalassemia. These studies
were designed with different objectives from the principles described
above, but demonstrated the safety (no significant severe side effects)
of iron chelation during the peri-transplant phase.[28]
Fritsch et al utilized Deferasirox during the administration of
conditioning regimens and it was found to be safe and reduced the
appearance of LPI shortly after allo-HSCT in this preliminary study.[29]
The
basic idea today would be to suppress, by adequate (not necessarily
intensive) peri-transplant chelation, the NTBI/LPI increment occurring
during transplant. Obviously, this rationale should be applied,
at the moment, only in the context of a well-designed controlled
clinical trial.
Free Iron Toxicity During the Early Transplant Phase (Before a Sustained Engraftment is Achieved)
Tissues
that have been damaged from iron toxicity before and during HCT
gradually restore their functionality. Nevertheless, it is believed
that, in some instances, ROS exposition persists also after transplant.
In vitro tests on cardiac and endocrine cells have shown how after a
prolonged and constant LPI exposition iron accumulates in organelles
and increases ROS formation, affecting major cell functions such as
permselectivity, electron transport activity, and viability. Again,
iron chelation with Deferasirox was effective in reducing iron-induced
cell damage and increasing viability of these cells.[30]
This
model could also be applied after a sustained engraftment, not
requiring blood transfusion, has been achieved. In fact, hematopoietic
cell transplantation does not eliminate the iron excess acquired during
previous years of thalassemia. Serum ferritin and the transferrin
saturation slowly return to normal levels and only in patients with a
very low iron burden before transplantation. [31]
Accordingly,
with these observations, excess iron removing is essential after HCT.
This recommendation is based on the evidence that progression of
liver disease to cirrhosis has been documented in some patients in the
years after transplantation. Data demonstrated a synergistic
deleterious effect of Hepatitis C Virus (HCV) infection with iron
overload with a multiplicative fibrogenic effect.[5,32]
Recently a basic research paper has been published demonstrating a
novel and intricate mechanism by which HCV interferes with the
crosstalk between the Nrf2/ARE-signaling, elevated ROS levels and
autophagy. Basically HCV impairing Nrf2/ARE-signaling through the ROS
increase create an amplified and deleterious hepatotoxic effect,
favoring the HCV autophagy and its release and spread, worsening the
preexistent hepatic failure.[33] Others synergisms with others damage factors (for example alcohol abuse) are likely.
Because
a condition of iron overload leads to the release of reactive oxygen
species, even in absence of ongoing transfusion need, defining iron
overload post-transplant is essential and the gold standard for iron
detection remain the liver iron concentration (LIC) because serum
ferritin is particularly unreliable in this setting.
Recently
the importance of LIC measurement by magnetic resonance imaging (MRI)
has grown since it is non-invasive, rapid, and widely available. Today
MRI techniques T2* and R2 are reported to have sensitivity and
specificity of 89% and 80% in determination of LIC, respectively.[34]
Few
studies examine the alterations of hepatic and myocardial T2* MRI
values in TDT patients after HCT just before starting chelation
therapy. The main study included fifty-two TDT patients with mean age
of 7.6 years. Hepatic and myocardial T2* values before and 6 months
after HCT were measured and analyzed. Results showed that there
was not a statistically significant increase in myocardial T2* values
after HCT, instead Hepatic T2* values significantly decreased after HCT
showing an increase of the liver iron.[35]
Serum
ferritin levels appeared to have a poor correlation with LIC in
thalassemic patients after HCT. Ferritin can be a good screening test
but a poor predictor of tissue iron overload.[36]
The
above reported ALLIVE study in malignancies has been shown that
only eLPI correlated with LIC (not ferritin neither hepcidin values)
and patients with elevated eLPI baseline values had a worsening outcome
due to an increased non -transplant related mortality.[17]
The
hypothesis is that LPI/ NTBI might be used to assess the iron
exposition status of organs after transplant, deciding initiation and
duration of iron chelation and monitoring efficacy with the goals to
protect organs from ROS exposition suppressing LPI and NTBI.
It
is important to note that, in the current state of knowledge, it must
be assumed that a normal transferrin saturation (i.e. 20-30%) excludes
the presence of toxic iron forms of in the circulation and consequent
progressing tissue damage; for this reason, it can be used as surrogate
of normal ROS level.
Iron Chelation Therapy after Transplant in TDT
Because
of the presence of effective erythropoiesis acquired by
transplantation, phlebotomy is the preferred mechanism to remove excess
iron after HCT. Phlebotomy is safe, inexpensive, and highly efficient.
It can be started once engraftment is sustained and preferably after
immunosuppressive therapy ending. Clinical improvement in liver and
cardiac function has been demonstrated after iron depletion by
phlebotomy in several instances.[37,38]
In
transplanted patients undergoing phlebotomy, iron can be completely
removed (final target: normal transferrin saturation, serum ferritin
concentration <100 μg/L), and after this achievement, patients are
free from iron overload and no maintenance therapy is required.
Duration of treatment is strictly related to the magnitude of the iron
overload, and it ranges from a few months to several years.
Two oral iron chelators have been tested in ß-TM (Deferiprone[39] and Deferasirox,[40]
but only Deferasirox has been tested after transplantation. Reported
cases of deferiprone-induced neutropenia in medically treated
thalassemia patients raise concern for its use in the post-transplant
setting.
The oral iron chelator Deferasirox has recently been
tested in this setting on patients in prospective trials. Firstly, data
from a prospective phase IV trials conducted by Vallejo et al showed
that Deferasirox is efficient and safe in the post-transplant period in
hematologic malignancies. Patients at least six months
post-transplanted were treated with deferasirox dispersible tablets
(DT) at a starting dose of 10 mg/kg/day for 52 weeks or until serum
ferritin was less than 400 ng/mL on two consecutive occasions. A
significant reduction from baseline in median serum ferritin and in
liver iron concentration at 52 weeks was observed in the overall
population. There were no drug-related serious adverse events.[41]
Similar
results are recently described in the Thalassemic population. A
prospective, phase II, multicenter, single-arm study evaluates the
efficacy and safety of deferasirox-DT in patients age >2 to <18
years with TDT who had undergone HCT and had evidence of iron overload
(serum ferritin >1000 µg/L; cardiac MRI T2* <20 ms, or liver iron
concentration ≥5 mg/g). Patients received deferasirox at an initial
dose of 10 mg/kg/day, with up-titration to a maximum of 20 mg/kg/day.
There was a continuous decrease in median serum ferritin level from
1718.0 µg/L at baseline to 845.3 µg/L following 52 weeks of therapy
(P < .001). There was also a significant decrease in median LIC and
an increase in median cardiac T2* from baseline to week 52. A
manageable safety profile was observed.[42]
A
prospective randomized 1 year phase II trial comparing efficacy
and safety of Deferasirox-DT versus the standard of care phlebotomy has
been recently published in the population of transplanted thalassemia
patients.[43] Deferasirox-DT starting dose was 10
mg/Kg/day increased till 20 mg/Kg/day during the trials. They showed
that Deferasirox is efficient and safe post-transplant; no differences
were reported in reducing serum ferritin ability in both arms.
The
advantage of Deferasirox is that it is administrated orally. The
disadvantages regard the possible renal and hepatic toxicity
considering that such patients are receiving in this period
cyclosporine and other drugs as well as its noticeable cost. In
2016 Jaekel et al showed that Deferasirox is efficient and safe, even
in patients receiving cyclosporine. Deferasirox was initiated at a
median of 168 days after HCT, with 84% of patients still on
immunosuppression. The incidence of AEs appeared to be dose related,
with 7.5 mg/kg/day of deferasirox-DT being the best-tolerated dose.
They concluded that low-dose deferasirox is an effective chelation
therapy after allogeneic HCT, with a manageable safety profile, even in
patients receiving cyclosporine.[44] The recently released film coated tablets (FCT) formulation promises to further increase tolerability.[45]
Reasons to select phlebotomies or deferasirox have been published.[13]
It
is important to note that all transplanted patients after successful
HCT face a normal life expectancy and therefore their target iron level
must be a “normal “iron burden with normal ferritin and, mostly, normal
transferrin saturation.
Conclusions
Reinterpreting
transplant predictive factors in the light of the current advances in
understanding iron homeostasis further supports the concept that the
key to successful transplantation in thalassemia is regular and
life‐long chelation therapy to consistently suppress tissue reactive
iron species and prevent tissue damage in the years before HCT. In the
next near future, the suppression of the free iron forms (LPI, NTBI and
ROS) could improve organ damage that could be important for the HCT and
possibly even the gene therapy outcome.
References
- Olivieri NF, Nathan DG, MacMillan JH, et al (1994)
Survival in medically treated patients with homozygous
beta-thalassemia. N Engl J Med 331:574-578. https://doi.org/10.1056/NEJM199409013310903
- Angelucci E (2010) Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program 2010:456-462. https://doi.org/10.1182/asheducation-2010.1.456
- Finotti A, Breda L, Lederer CW, et al (2015) Recent trends in the gene therapy of β-thalassemia. J Blood Med 6:69-85. https://doi.org/10.2147/JBM.S46256
- Armand
P, Kim HT, Cutler CS, et al (2007) Prognostic impact of elevated
pretransplantation serum ferritin in patients undergoing myeloablative
stem cell transplantation. Blood 109:4586-4588. https://doi.org/10.1182/blood-2006-10-054924
- Pullarkat
V, Blanchard S, Tegtmeier B, et al (2008) Iron overload adversely
affects outcome of allogeneic hematopoietic cell transplantation. Bone
Marrow Transplant 42:799-805. https://doi.org/10.1038/bmt.2008.262
- Schaible UE, Kaufmann SHE (2004) Iron and microbial infection. Nat Rev Microbiol 2:946-953. https://doi.org/10.1038/nrmicro1046
- Pilo F, Angelucci E (2017) A storm in the niche: Iron, oxidative stress and haemopoiesis. Blood Rev. https://doi.org/10.1016/j.blre.2017.08.005
- Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of Mammalian iron metabolism. Cell 142:24-38. https://doi.org/10.1016/j.cell.2010.06.028
- Sajadimajd S, Khazaei M (2018) Oxidative Stress and Cancer: The Role of Nrf2. Curr Cancer Drug Targets 18:538-557. https://doi.org/10.2174/1568009617666171002144228
- Coates TD (2014) Physiology and pathophysiology of iron in hemoglobin-associated diseases. Free Radic Biol Med 72:23-40. https://doi.org/10.1016/j.freeradbiomed.2014.03.039
- Lucarelli
G, Galimberti M, Polchi P, et al (1990) Bone marrow transplantation in
patients with thalassemia. N Engl J Med 322:417-421. https://doi.org/10.1056/NEJM199002153220701
- Angelucci
E, Pilo F, Coates TD (2017) Transplantation in thalassemia: Revisiting
the Pesaro risk factors 25 years later. Am J Hematol 92:411-413. https://doi.org/10.1002/ajh.24674
- Angelucci
E, Pilo F (2016) Management of iron overload before, during, and after
hematopoietic stem cell transplantation for thalassemia major. Ann N Y
Acad Sci 1368:115-121. https://doi.org/10.1111/nyas.13027
- Alessandrino
EP, Porta Della MG, Bacigalupo A, et al (2010) Prognostic impact of
pre-transplantation transfusion history and secondary iron overload in
patients with myelodysplastic syndrome undergoing allogeneic stem cell
transplantation: a GITMO study. Haematologica 95:476-484. https://doi.org/10.3324/haematol.2009.011429
- Platzbecker
U, Bornhäuser M, Germing U, et al (2008) Red blood cell transfusion
dependence and outcome after allogeneic peripheral blood stem cell
transplantation in patients with de novo myelodysplastic syndrome
(MDS). Biol Blood Marrow Transplant 14:1217-1225. https://doi.org/10.1016/j.bbmt.2008.08.006
- Kataoka
K, Nannya Y, Hangaishi A, et al (2009) Influence of pretransplantation
serum ferritin on nonrelapse mortality after myeloablative and
nonmyeloablative allogeneic hematopoietic stem cell transplantation.
Biol Blood Marrow Transplant 15:195-204. https://doi.org/10.1016/j.bbmt.2008.11.012
- Wermke
M, Eckoldt J, Götze KS, et al (2018) Enhanced labile plasma iron and
outcome in acute myeloid leukaemia and myelodysplastic syndrome after
allogeneic haemopoietic cell transplantation (ALLIVE): a prospective,
multicentre, observational trial. Lancet Haematol 5:e201-e210. https://doi.org/10.1016/S2352-3026(18)30036-X
- Pullarkat
V (2014) Iron toxicity in hematopoietic stem cell transplantation:
Strike while the iron is labile. Acta Haematol 131:220-221. https://doi.org/10.1159/000355827
- Naoum
FA, Espósito BP, Ruiz LP, et al (2014) Assessment of labile plasma iron
in patients who undergo hematopoietic stem cell transplantation. Acta
Haematol 131:222-226. https://doi.org/10.1159/000355192
- Kautz
L, Jung G, Valore EV, et al (2014) Identification of erythroferrone as
an erythroid regulator of iron metabolism. Nat Genet 46:678-684. https://doi.org/10.1038/ng.2996
- Naoum
FA, Espósito BP, Cançado RD (2016) Impact of conditioning and
engraftment on iron status in hematopoietic stem cell transplantation:
Contribution of labile plasma iron. Hematol Oncol Stem Cell Ther
9:165-167. https://doi.org/10.1016/j.hemonc.2016.07.001
- Duca
L, Cappellini MD, Baronciani D, et al (2018) Non-transferrin-bound iron
and oxidative stress during allogeneic hemopoietic stem cell
transplantation in patients with or without iron overload. Am J Hematol
93:E250-E252. https://doi.org/10.1002/ajh.25201
- Bigarella CL, Liang R, Ghaffari S (2014) Stem cells and the impact of ROS signaling. Development 141:4206-4218. https://doi.org/10.1242/dev.107086
- Ludin
A, Gur-Cohen S, Golan K, et al (2014) Reactive oxygen species regulate
hematopoietic stem cell self-renewal, migration and development, as
well as their bone marrow microenvironment. Antioxid Redox Signal
21:1605-1619. https://doi.org/10.1089/ars.2014.5941
- Chai
X, Li D, Cao X, et al (2015) ROS-mediated iron overload injures the
hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor
cells in mice. Sci Rep 5:10181. https://doi.org/10.1038/srep10181
- Okabe
H, Suzuki T, Uehara E, et al (2014) The bone marrow hematopoietic
microenvironment is impaired in iron-overloaded mice. Eur J Haematol
93:118-128. https://doi.org/10.1111/ejh.12309
- Zhang
Y, Zhai W, Zhao M, et al (2015) Effects of iron overload on the bone
marrow microenvironment in mice. PLoS ONE 10:e0120219. https://doi.org/10.1371/journal.pone.0120219
- Gaziev
D, Giardini C, Angelucci E, et al (1995) Intravenous chelation therapy
during transplantation for thalassemia. Haematologica 80:300-304.
PMid:7590497
- Fritsch A, Langebrake C,
Nielsen P, et al (2011) Deferasirox (Exjade®) Given During Conditioning
Regimen (FLAMSA/Busulfan/ATG) Reduces the Appearance of Labile Plasma
Iron in Patients Undergoing Allogeneic Stem Cell Transplantation. Blood
118:3023.
- Naoum FA, Espósito BP,
Zotarelli Filho IJ (2017) Impact of labile plasma iron and iron
chelation on the viability of cultured mononuclear cells from patients
undergoing autologous hematopoietic stem cell transplantation. Blood
Res 52:135-136. https://doi.org/10.5045/br.2017.52.2.135
- Lucarelli
G, Angelucci E, Giardini C, et al (1993) Fate of iron stores in
thalassaemia after bone-marrow transplantation. Lancet 342:1388-1391. https://doi.org/10.1016/0140-6736(93)92753-G
- Angelucci
E, Muretto P, Nicolucci A, et al (2002) Effects of iron overload and
hepatitis C virus positivity in determining progression of liver
fibrosis in thalassemia following bone marrow transplantation. Blood
100:17-21. https://doi.org/10.1182/blood.V100.1.17 PMid:12070002
- Medvedev
R, Ploen D, Spengler C, et al (2017) HCV-induced oxidative stress by
inhibition of Nrf2 triggers autophagy and favors release of viral
particles. Free Radic Biol Med 110:300-315. https://doi.org/10.1016/j.freeradbiomed.2017.06.021
- Atilla
E, Toprak SK, Demirer T (2017) Current Review of Iron Overload and
Related Complications in Hematopoietic Stem Cell Transplantation. Turk
J Haematol 34:1-9. https://doi.org/10.4274/tjh.2016.0450
- Hamidieh
AA, Tayebi S, Moeininia F, et al (2014) T2(∗) MRI changes in the heart
and liver of ex-thalassemic patients after hematopoietic stem cell
transplantation. Hematol Oncol Stem Cell Ther 7:103-108. https://doi.org/10.1016/j.hemonc.2014.06.002
- Jarisch
A. Salzmann-Manrique E, Cario H. et al. Serum ferritin is not a
reliable predictor to determine iron overload in thalassemia major
patients post-hematopoietic stem cell transplantation. Eur J Haematol
2018 Dec;101(6): 791-797 https://doi.org/10.1111/ejh.13169 PMid:30187571
- Angelucci
E, Muretto P, Lucarelli G, et al (1998) Treatment of iron overload in
the "ex-thalassemic." Report from the phlebotomy program. Ann N Y Acad
Sci 850:288-293. https://doi.org/10.1111/j.1749-6632.1998.tb10485.x PMid:9668550
- Mariotti
E, Angelucci E, Agostini A, et al (1998) Evaluation of cardiac status
in iron-loaded thalassaemia patients following bone marrow
transplantation: improvement in cardiac function during reduction in
body iron burden. Br J Haematol 103:916-921. https://doi.org/10.1046/j.1365-2141.1998.01099.x PMid:9886301
- Belmont
A, Kwiatkowski JL (2017) Deferiprone for the treatment of transfusional
iron overload in thalassemia. Expert Rev Hematol 10:493-503 https://doi.org/10.1080/17474086.2017.1318052
- Galanello
R, Campus S, Origa R (2012) Deferasirox: pharmacokinetics and clinical
experience. Expert Opin Drug Metab Toxicol 8:123-134. https://doi.org/10.1517/17425255.2012.640674
- Vallejo
C, Batlle M, Vázquez L, et al (2014) Phase IV open-label study of the
efficacy and safety of deferasirox after allogeneic stem cell
transplantation. Haematologica 99:1632-1637. https://doi.org/10.3324/haematol.2014.105908
- Yesilipek
MA, Karasu G, Kaya Z, et al (2018) A Phase II, Multicenter, Single-Arm
Study to Evaluate the Safety and Efficacy of Deferasirox after
Hematopoietic Stem Cell Transplantation in Children with β-Thalassemia
Major. Biol Blood Marrow Transplant 24:613-618. https://doi.org/10.1016/j.bbmt.2017.11.006
- Inati
A, Kahale M, Sbeiti N, et al (2017) One-year results from a prospective
randomized trial comparing phlebotomy with deferasirox for the
treatment of iron overload in pediatric patients with thalassemia major
following curative stem cell transplantation. Pediatr Blood Cancer
64:188-196. https://doi.org/10.1002/pbc.26213
- Jaekel
N, Lieder K, Albrecht S, et al (2016) Efficacy and safety of
deferasirox in non-thalassemic patients with elevated ferritin levels
after allogeneic hematopoietic stem cell transplantation. Bone Marrow
Transplant 51:89-95. https://doi.org/10.1038/bmt.2015.204
- Taher
AT, Origa R, Perrotta S, et al (2017) New film-coated tablet
formulation of deferasirox is well tolerated in patients with
thalassemia or lower-risk MDS: Results of the randomized, phase II
ECLIPSE study. Am J Hematol 92:420-428. https://doi.org/10.1002/ajh.24668
[TOP]