Tuysuz
G.1, Yildiz I.1,
Ozdemir N.1, Adaletli İ.2,
Kurugoglu S.2, Apak H.1,
Dervisoglu S.3, Bozkurt S.4
and Celkan T.1.
1
İstanbul University Cerrahpasa Medical Faculty Pediatric Hematology and
Oncology Dept.
2 İstanbul University Cerrahpasa Medical Faculty
Radiology Dept.
3 İstanbul University Cerrahpasa Medical Faculty
Pathology Dept.
4 Akdeniz University Medical Faculty Statistics
Dept.
Correspondence to:
Gulen Tuysuz. Istanbul University Cerrahpasa
Medical Faculty Pediatric Hematology and Oncology Dept. Adress: Koca
Mustafa Paşa Mahallesi, Cerrahpaşa Caddesi No:53, 34096
Fatih/İstanbul-TURKEY. Tel: +905053159667, Fax: +902126328633. E-mail:
gulentuysuz@hotmail.com
Published: May 1, 2019
Received: November 9, 2018
Accepted: April 17, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019035 DOI
10.4084/MJHID.2019.035
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Objectives:
To review a single center outcome of patients with Langerhans Cell
Histiocytosis diagnosed at a tertiary referral hospital from Turkey.
Methods:
The files between 1989 and 2015 of 80 patients with LCH were
retrospectively analyzed.
Results:
During the 25 years, 80 patients were diagnosed with LCH. The median
age at diagnosis was 53 months (2-180 months) and the median follow-up
time of patients was 10 years and 9 months (24 months-25 years). Bone
was the most frequently affected organ (n:60, 75%). Initially, 43
patients (54%) had single system (SS) disease, 20 patients (25%) had
multisystem (MS) disease without risk organ involvement (MS-RO-), and
17 patients (21%) had a multisystem disease with risk-organ involvement
(MS-RO+). The overall survival (OS) rate was 91%, and event-free
survival (EFS) rate was 67% at 10 years. 10-year OS rate was lower for
patients with MS-RO+ (65%) when compared to those with, MS-RO-, and SS
(100%, 97%, p value=<0.001). The overall survival rate was also
lower in patients with lack of response to systemic chemotherapy on
12th week (p=<0.001), younger age (<2 years) at
presentation
(p=<0.02), skin involvement (<0.001) and lack of bone
lesions at
presentation (<0.001).
Discussion:
In the group with MS-RO+, OS is significantly low compared to other
groups. Further efforts are warranted to improve survival in MS-RO+
patients.
|
Introduction
Langerhans
cell histiocytosis (LCH) is a rare neoplasm caused by an abnormal
oligoclonal proliferation of Langerhans cells and their accumulation at
various tissues and organs.[1,2,3]
The overall incidence rate for LCH
was reported as 2.6 cases per million child years, and males are
affected to a higher degree than females.[4]
Langerhans cell
histiocytosis is categorized into two major categories based on the
extent of disease:[5] single-system
(SS) and multisystem (MS). The
clinical presentation and outcome of the disease are diverse, and
treatment options differ according to the extent and severity of
disease.[6-8]
In this article, we present a retrospective analysis
of LCH cases diagnosed at a tertiary referral hospital in Turkey over
the past 25 years. Our aim was to describe the course of the disease
and evaluate the factors that have an effect over survival in our
cohort.
Materials and Methods
The
data of patients with LCH, treated at Istanbul University, Cerrahpasa
Medical School Hospital Pediatric Hematology-Oncology Department
between 1989 and 2015, were retrospectively analyzed from the medical
records. A total of 80 patients were included. Their files were
reviewed for demographic characteristics, clinicopathological features,
laboratory findings, treatment regimens, and outcome.
Disease
staging and organ dysfunctions were evaluated by disease history,
physical examination, laboratory tests, and imaging studies. Complete
blood count, liver and kidney function tests, serum electrolytes,
ferritin, total bilirubin, PT, a PTT, urine osmolality were checked in
all patients. Bone marrow aspirate and biopsy were performed only in
multisystem patients. The radiological examination included at least
chest X-ray and skeletal radiograph survey. Other further imaging
modalities such as ultrasonography, computerized tomography, magnetic
resonance imagining, bone scintigraphy, positron emission tomography
and pulmonary function tests were performed when evaluation of the
extent of disease was required.
LCH can be clinically be
classified in two general groups, single system and multisystem. Risk
organ involvement in the multisystem group is considered to be the most
aggressive form of the disease.[4,5]
While preparing the manuscript for
publication, patients were retrospectively staged according to
currently ongoing LCV IV trial of Histiocyte Society[9]
even though
different protocols have been used for classification and treatment of
these patients. According to LCH IV trial, in monosystemic (single
system) form, one organ or system is involved; such as bone (either as
a single bone; monosystem unifocal bone or more than one bone;
monosystem multifocal bone), skin or lymph nodes. In multisystemic form
two or more organs or systems are involved; either with or without risk
organs (hematopoietic system involvement, spleen and liver). Different
from the previous protocols, the lung is not considered as a risk organ
in LCH IV trial.
Treatment included local steroid therapy,
radiotherapy, chemotherapy, surgical excision of the lesion (even
though it is not recommended any longer), or a combination of these
modalities. Depending on the year of admission, patients were treated
by DAL-HX 83 protocol[10,11,12]
between 1989 and 1991, LCH-1
protocol[12] between 1992 and
1996, LCH-II protocol[7] between
1996 and
2004 and LCH-III protocol[13]
after 2004.
For the evaluation of
response, the response criteria of LCH-1 Study[6]
were employed.
Responders had a complete resolution (NAD) or continuous regression of
disease; intermediate responders were patients with active disease who
had either stable disease or a mixed response (a regression of disease
but the appearance of new lesions in another site or organ system), and
non-responders had progression of the disease. Reactivation was defined
as a reappearance of disease signs or symptoms after complete disease
resolution.[11]
Statistics
Continuous
variables are presented as median (mean-max) deviation, while
categorical variables are given as percentages. The Shapiro Wilk test
was used to verify the normality of the distribution of continuous
variables. Statistical analysis of clinical characteristics between two
groups consisted of unpaired t-tests for parametric data and Mann
Whitney U test, whereas the chi-square/Fisher's exact tests were used
for categorical variables. The Kaplan-Meier method was used to estimate
survival as a function of time, and the log-rank test analyzed survival
differences. Analyses were performed with PASW 18 (SPSS/IBM, Chicago,
IL, USA) software and two-tailed P value less than 0.05 was considered
statistically significant.
Results
Patient
Characteristics.
Among 80 eligible patients with LCH, 56 of them were male, and 24 were
female (M/F: 2.3). Median age at diagnosis was 36 months (2 months to
15 years). Patients in the SS group had a higher median age at
diagnosis when compared to MS-RO-
and MS-RO+
groups (p=0.01 and
p=0.0001 respectively). General characteristics of patients are shown
in Table 1.
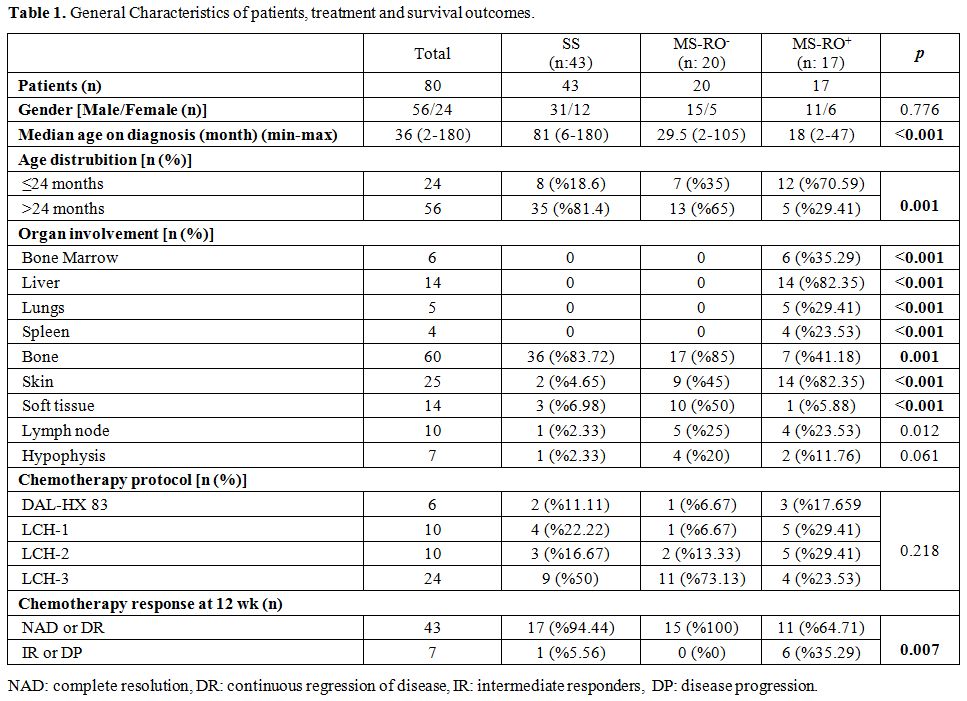 |
Table
1. General Characteristics of patients, treatment and survival outcomes. |
Initial
symptoms.
At the time of diagnosis, swelling (n: 33, 41%) was the most recorded
referral symptom followed by pain (n: 24, 30%) in which 21 were related
to bone and 7 presented with limping gait. Skin rash or eruption was
noted in 16 (20%) of the patients while polyuria and polydipsia was the
presenting symptom in 7 (8.7%) of the patients.
Physical examination and organ involvement.
The most frequently affected organ was bone (n: 60, 75%). In MS-RO+
group, besides risk organ infiltration, skin involvement was also
statistically higher (n:14; p=0.0001) compared to the other two groups.
Bone involvement was statistically high in the SS group (n: 36;
p=0.01), and soft tissue involvement was statistically higher in MS-RO-
group (n:10; p=0.0001). Distribution of organ involvement varied
significantly by patient age. Status of patients according to age is
illustrated in Table 2.
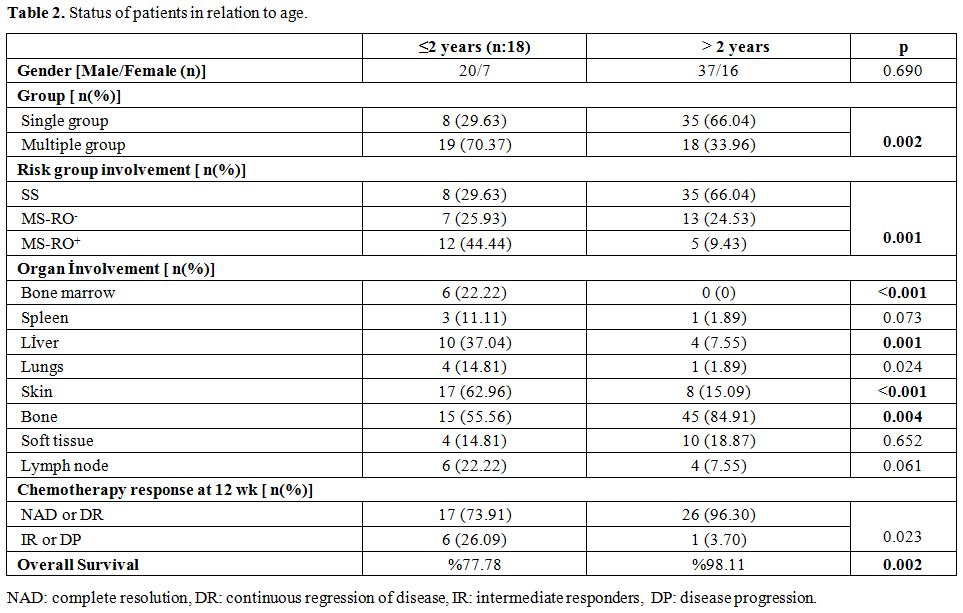 |
Table
2. Status of patients in relation to age. |
Diagnosis.
Among 80 patients enrolled to study, 76 (95%) had a histological
diagnosis of LCH based on characteristics histological appearance of
LCH lesions on hematoxylin and eosin and positive immunohistochemical
staining with CD1a and/or S-100. The diagnosis was based on
radiological and clinical findings in 4 (5%) patients, because of the
surgical risk due to localization in the paravertebral area.
Staging.
Forty-three patients (53.75%) presented with SS disease and 37 patients
(46.25%) presented with multisystem disease. Distribution of risk
groups varied according to the age as is shown in Table 2.
Treatment. Patients
were treated according to the extent of the disease. Details of
treatment regimens are illustrated in Table 3.
Among 22 patients with unifocal bone lesions 16 were treated with
surgical curettage/excision and the rest 6, who had involvement of
weight-bearing bones, skull base, temporal bones and vertebral column,
were treated with chemotherapy (n:5) and local radiotherapy (n:1). For
the patients in the multisystem low-risk group (n: 20), 15 were treated
with chemotherapy, and the rest 5 were treated with combination
treatments. All patients in the multisystem high risk group (n: 17)
were treated with systemic chemotherapy. A total of 50 patients from
all groups received chemotherapy as the treatment. According to
chemotherapy response on week 12, 43 patients (86%) were classified as
responders; of these, 33 (66%) had NAD, and 10 (20%) had DR. Seven
patients (14%) were evaluated as non-responders; of these 3 (6%) had IR
and 4 (8%) had DP. Chemotherapy response was statistically worse in
MS-RO+
group compared to the other two groups (p=0.007) and in patients, under
2 years of age (p=0.023) as is shown in Table 1 and 2.
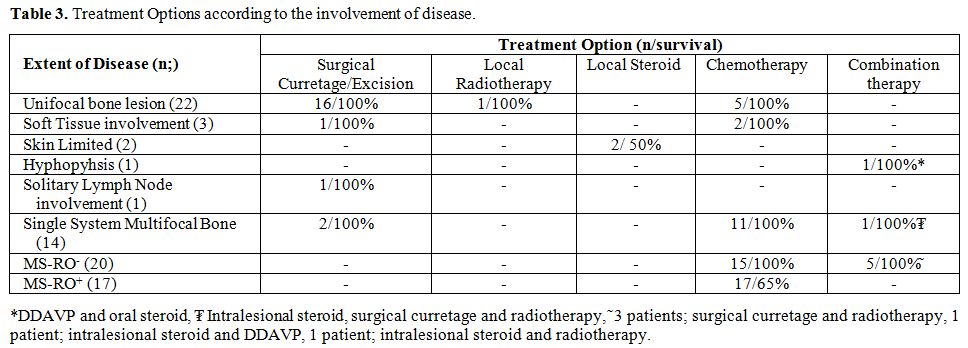 |
Table
3. Treatment Options according to the involvement of disease. |
Treatment
response and outcome.
The median follow up time was 10 years and 9 months (1 month to 25
years), 10-year overall survival (OS) rate in the entire patient cohort
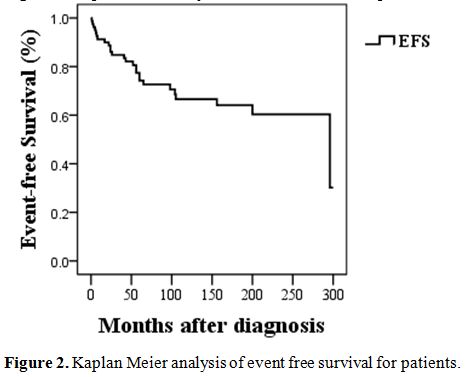
were 91.25 %, and EFS (event-free survival) rate was 67.5%. Seven
patients died. One patient in the single system group (skin
involvement) has developed reactivation of risk organ involvement
during follow up and was lost due to progressive disease. Besides this
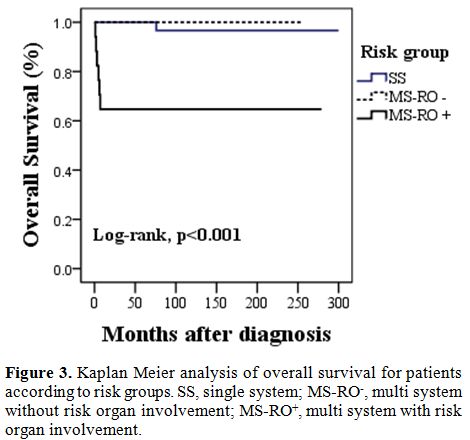
patient, the rest 6 were in MS-RO+ group. 10-year
OS rate was lower for patients with MS-RO+
(65%) when compared to those with, MS-RO-,
and SS (100%, 97%, p value=<0.001).
Regarding
the age of patients' OS rate at 10 years from diagnosis was 77% for
patients younger than 2 years of age and 98% for patients older than 2
years of age (p=0.02). Bone involvement was reported in 60 patients
(75%). Ten year OS rate was significantly higher in patients with bone
involvement than in those with extraosseous disease site involvement
(100% vs. 65%; p=<0.001). Even among patients of MS-RO+
group, presence of bone lesions was associated with better OS (%100 vs.
% 40; p=0.016). Ten year OS rate was significantly higher in patients
who responded to initial treatment at 12 weeks compared to those who
did not (100% vs. 14%; p=<0.001) and also in patients with skin
involvement as is shown in Table
4. Due to the difference in the distribution of deaths
among groups, we could not perform multivariate analysis.
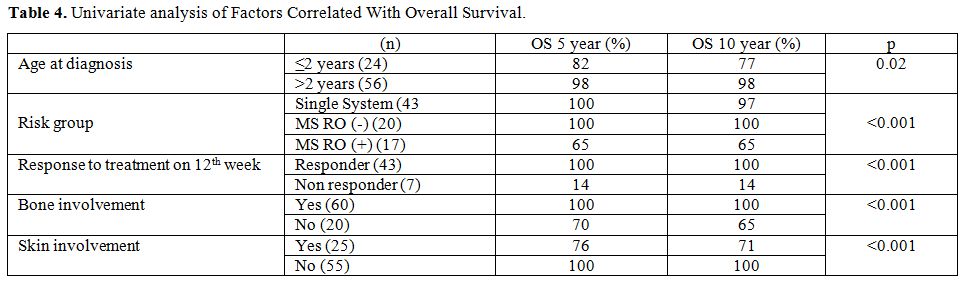 |
Table
4. Univariate analysis of Factors Correlated With Overall
Survival. |
Reactivation.
Out of 80 patients, 20 (25%) experienced at least one reactivation. The
first reactivation occurred 2-46 months (median: 11 mo) after the
initial diagnosis. Regarding the timing, one patient (5%) reactivated
during induction treatment, 4 (20%) patients reactivated during
continuation treatment and 15 patients (75%) reactivated while on
follow up. Among these reactivations, 4 occurred in the first year and
the rest 11 afterward (2-46 months, median: 17 months after the initial
diagnose). Among the patients with first reactivation, 7 patients had
SS MFB, 6 patients had SS SS (single system, single side), 4 patients
had MS-RO+,
and 3 patients had MS-RO-
disease. The most clinical pattern of reactivation was limited to the
bone. Bone reactivation was observed in 16 of the 20 patients (7
patients unifocal, 4 patients multifocal bone and in 5 patients
reactivation was associated with other organs' involvement). Risk organ
reactivation was observed in only 3 patients (15%). Patients with
reactivated disease were treated with chemotherapy (n:17), or local
therapy (radiotherapy (n:1), curettage (n:1) and intralesional steroid
(n:1). Five patients experienced multiple reactivations: one patient
experienced 2, three patients experienced 3, and one patient
experienced 4 reactivations in the follow-up; within a mean period of
17 (9-26 mo) months. The 10-year reactivation rates for SS-SS, MS-RO-, and MS-RO+
patients were 30%, 15%, and 23.5% respectively. Reactivation rate did
not differ statistically according to the involved organs or risk
groups. Reactivation did not affect mortality except for one patient in
group SS-SS (MFB) who relapsed in follow-up with risk-organ involvement
and died due to disease progression despite the combined chemotherapy
protocols in 6 months. The EFS of the cohort is shown in Figure 2.
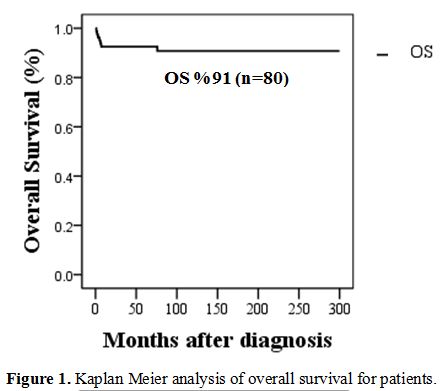 |
Figure 1. Kaplan
Meier analysis of overall survival for patients. |
 |
Figure
2. Kaplan Meier analysis of event free survival for patients. |
 |
Figure
3. Kaplan Meier
analysis of overall survival for patients according to risk groups. SS,
single system; MS-RO-, multi system without risk organ involvement;
MS-RO+, multi system with risk organ involvement. |
Discussion
Because
LCH is a rare disease, disease-related publications in the literature
are usually multi-institutional. This report is one of the rare
single-center studies within 25 years including 80 pediatric patients
with sufficient follow up duration. Our aim was to describe the course
of the disease and evaluate the outcomes.
The demographic features
of LCH patients in our cohort were comparable with previous reports
showing early onset of disease and male predominance.[14,15]
The median age of our patients at diagnosis was 36 months; more than
1/3 of patients were under 2 years, and the male/female ratio was 2.3.
In
concordance with previous reports, the majority of our patients
presented with symptoms related to bone (71%) including swelling, pain
and limping gait.[16,17,18] The
most affected organs
by disease in our study after bone were skin, soft tissue and liver
retrospectively and which was also in line with the previously reported
series in the literature.[17,18]
Presence of bone lesions at diagnosis was associated with better OS in
our cohort as was described in the literature before.[19,20]
Even among MS-RO+
group, OS was significantly better in patients with bone disease (%100
in the presence of bone disease and 40% in the absence of bone disease;
p=0.016). Even though this is in concordance with previous reports
showing the favorable course of patients with bone involvement among
MS-RO+ patients, in
our opinion, our number of patients is too low (n:17) to contribute to
this hypothesis.[20]
In
our cohort, group involvement differed according to age, while the
children older than 2 years mostly presented with SS disease and bone
involvement, the younger group (≤2 years of age) presented with more
multisystem disease, skin and risk organ involvement. This was in
concordance with previous studies in the literature.[16,17,19]
Kim
et al. described "bone" as the most common site of involvement in their
study among the patients between 1 to 5 years of age.[16,21]
In 2012, Postini et al. reported their 40 years of experience with
pediatric LCH patients. Single system unifocal bone involvement was the
most observed clinical presentation in patients over 2 years of age.[20,22]
In a nationwide survey from Korea, young age at diagnosis (<2
years)
was associated with multisystem risk organ involvement resulting in
higher mortality.[16]
In LCH, the course of the
disease is highly heterogeneous and it is related to the extent of
organ involvement. In 2014, Lee at al reported the outcome of 22 years’
experience. The OS rate was significantly low in patients with
risk-organ involvement.[21] In the
study by Yagci et
al. where the outcome of 217 LCH patients was described OS and EFS
rates were significantly worse in MS-RO+.[17] In our study, patients with SS
disease and MS-RO-
had excellent survival rates. All patients except one survived in these
two groups. The only patient dead was a boy who had a reactivation of
risk organ involvement during follow up. Besides this patient, all the
other deaths were observed in the MS-RO+. Our findings
support the hypothesis that risk organ involvement is a strong negative
predictor of outcome in LCH patients.
Skin involvement was observed in 25 (31%) of our cohort. Age<2
years and MS-RO+ were
associated with skin involvement (p<0.001) as was shown in the
literature before.[21] Several
studies revealed the presence of somatic BRAF-V600E mutation on skin
biopsy.[23,24] The existence of
BRAF-V600E in circulating blood has been associated with disease
recurrence.[25]
In our cohort, a skin biopsy or peripheral blood were not available for
analyses of BRAF-V600E mutation. However, in univariate analyses
patients with skin involvement had lower EFS and OS. Due to the close
association of skin disease with risk-organ involvement and the low
number of patients enrolled we cannot conclude whether skin involvement
is an independent predictor of poor outcome. Prospective multicenter
trials are needed to determine the effect of skin involvement over the
outcome in LCH patients.
There is no current standard management
protocol for patients whose disease is unresponsive to frontline
therapy or who present with multiple reactivations. Even though
patients with single bone or low-risk multisystem reactivation respond
well to second-line treatments such as 6-mercaptopurine and
methotrexate, patients with risk-organ reactivation have inadequate
response even to salvage protocols. Treatment of refractory LCH
patients with 2CdA as monotherapy has shown a higher response rate in
patients with non-risk organs involvement, but limited activity in
refractory patients with risk-organ involvement.[26]
Combination of 2CdA with Cytarabine (Ara-C) as a salvage protocol has
promising results.[27] Even in the
MS-RO+ group, 5 year
survival rate was reported to be 85% in the phase II study by Donadeiu
et al.[28] The principal declared
side effect of this treatment was severe hematologic toxicity and
arising severe infection.[28]
Currently, ongoing prospective LCH-IV study is evaluating the effect of
2CdA and Ara-C combination chemotherapy for risk organ involved
refractory LCH patients.[9,27] In
our cohort we treated 2 of our patients with a combination of 2CdA and
Ara-C; one was a girl with single system bone involvement who relapsed
during maintenance therapy from multiple bones. She achieved remission
with 2CdA treatment until now. The second patient was initially staged
in single system group (skin involvement) but had reactivation of risk
organ involvement during follow up. He died because of progressive
disease despite 2CdA treatment. In our study, the number of patients
was too few to report 2CdA efficiency. In the literature, some case
reports are showing the efficacy of Clofarabine as monotherapy in
refractory LCH patients.[26]
Rodriguez-Galindo et al.
showed complete remission in 2 refractory LCH patients (both without
risk organ involvement) with Clofarabine therapy who were unresponsive
to 2-CdA treatment.[29] This
finding was in
concordance with the recent study showing the superiority of
Clofarabine treatment in non-risk organ involved refractory LCH
patients.[30]
There are also promising reports regarding the Lenalidomide plus
steroid treatment in refractory patients.[31,32]
The main advantage of this protocol is the feasibility of treatment at
an outpatient clinic, cost-affectivity of the drug and reported limited
toxicity. However, literature regarding this protocol is scarce in the
pediatric population.
After the description of recurrent oncogenic
mutations affecting the MAPK pathway in LCH patients, targeted
therapies such as BRAF, MEK or BRAF/MEK inhibitors were reported to be
useful for patients with these mutations who even were unresponsive to
salvage treatments.[22,33,34]
However, further studies are warranted to reveal the efficacy, safety,
and long term outcome in the pediatric population for targeted
therapies.
Reactivation is a common problem in the treatment of patients with LCH.[18,20,21]
The total reactivation rate in our cohort was 25%. This rate is similar
to the reported data.[16,20,25]
Reactivations predominantly affected the bones as was shown in the
literature before.[19,20]
Even in the group with multifocal bone involvement or in patients with
multiple reactivations, recurrence or the disease, were not associated
with mortality. Only one patient with MS-RO+
reactivation died despite rescue treatment, which suggests the severity
of risk organ involvement also in disease reactivation. The 5-year
reactivation rates of our patients did not differ between the groups,
which was contradictory to the previous reports in where higher
reactivation rates were reported in the MS group.[16]
In our opinion, this is related to poor outcome in MS-RO+
group. Because most of these patients (5 of 6) could not get into
remission, they died in early stages of treatment before developing any
reactivation.
Conclusions
In conclusion,
our study shows favorable disease course in SS and MS-RO-
groups in LCH patients. Patients within these groups, survive with
chemotherapy, even if they develop multiple reactivations. Risk organ
involvement, younger age at presentation (<2 years),
unresponsive to
induction treatment, skin involvement, and absence of bone involvement
at diagnosis remained subgroups of worse outcome in our cohort. Further
improvement with more potent agents especially during induction is
warranted for the treatment of this group.
References
- Willman CL, Busque L,
Griffith BB, et al.
Langerhans'-cell histiocytosis (histiocytosis X)-a clonal proliferative
disease. N Engl J Med 1994; 331: 154-160. https://doi.org/10.1056/NEJM199407213310303
PMid:8008029
- Nezelof
C, Basset F. An hypothesis Langerhans cell histiocytosis: the failure
of the immune system to switch from an innate to an adaptive mode.
Pediatr Blood Cancer 2004; 42: 398-400 https://doi.org/10.1002/pbc.10463
PMid:15049008
- Grois
N, Potschger U, Prosch H, et al. Risk factors for diabetes insipidus in
Langerhans cell histiocytosis. Pediatr Blood Cancer 2006; 46: 228-233https://doi.org/10.1002/pbc.20425.
PMid:16047354
- Alston
RD, Tatevossian RG, McNally RJ, et al. Incidence and survival of
childhood LCH in northwest England from 1954 to 1998. Pediatr Blood
Cancer 2007; 48: 555-60. https://doi.org/10.1002/pbc.20884
PMid:16652350
- Haupt
R, Minkov M, Astigarraga I, et al.; Euro Histio Network. Langerhans
cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up,
and treatment for patients till the age of 18 years. Pediatr Blood
Cancer 2013; 60: 175-84. https://doi.org/10.1002/pbc.24367
PMid:23109216 PMCid:PMC4557042
- Gadner
H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of
treatment for multisystem Langerhans' cell histiocytosis. J Pediatr
2001; 138: 728-34. https://doi.org/10.1067/mpd.2001.111331
PMid:11343051
- Gadner
H, Grois N, Pötschger U, et al; Histiocyte Society. Improved outcome in
multisystem Langerhans cell histiocytosis is associated with therapy
intensification. Blood 2008; 111: 2556-62. https://doi.org/10.1182/blood-2007-08-106211
PMid:18089850
- Kudo
K, Ohga S, Morimoto A, et al. Improved outcome of refractory Langerhans
cell histiocytosis in children with hematopoietic stem cell
transplantation in Japan. Bone Marrow Transplant 2010; 45: 901-6. https://doi.org/10.1038/bmt.2009.245
PMid:19767778
- LCH-IV,
International Colloborative Treatment Protocol for Children and
Adolescents with Langerhans Cell Histiocytosis. Amended protocol
version 1.3 Nov 25. 2015.
- Gadner H,
Heitger A, Grois N, et al. Treatment strategy for disseminated
Langerhans cell histiocytosis. DAL HX-83 Study Group. Med Pediatr Oncol
1994; 23: 72-80. https://doi.org/10.1002/mpo.2950230203
PMid:8202045
- Minkov
M, Grois N, Heitger A, et al. Treatment of multisystem Langerhans cell
histiocytosis. Results of the DAL-HX 83 and DAL-HX 90 studies. DAL-HX
Study Group. Klin Padiat 2000; 212: 139-44. https://doi.org/10.1055/s-2000-9667
PMid:10994540
- S.
Ladisch and H. Gadner. Treatment of Langerhans cell
histiocytosis--evolution and current approaches. Br J Cancer Suppl
1994; 23: 41-6.
- Ramzan M, Yadav SP.
Langerhans cell histiocytosis presenting as isolated mediastinal mass
in an infant. Indian Pediatr 2014; 51: 397-8. https://doi.org/10.1007/s13312-014-0405-0
PMid:24953585
- Martin
A, Macmillan S, Murphy D, Carachi R. Langerhans cell histiocytosis: 23
years' paediatric experience highlights severe long-term sequelae.
Scott Med J 2014; 59: 149-57. https://doi.org/10.1177/0036933014542387
PMid:24996784
- Babeto
LT, de Oliveira BM, de Castro LP, et al. Langerhans cell histiocytosis:
37 cases in a single brazilian institution. Rev Bras Hematol Hemoter
2011; 33: 353-7. https://doi.org/10.5581/1516-8484.20110098
PMid:23049339 PMCid:PMC3415777
- Kim
BE, Koh KN, Suh JK, et al; Korea Histiocytosis Working Party.Clinical
features and treatment outcomes of Langerhans cell histiocytosis: a
nationwide survey from Korea histiocytosis working party. J Pediatr
Hematol Oncol 2014; 36: 125-33. https://doi.org/10.1097/MPH.0000000000000054
PMid:24276037
- Yağci
B, Varan A, Cağlar M, et al. Langerhans cell histiocytosis:
retrospective analysis of 217 cases in a single center. Pediatr Hematol
Oncol 2008; 25: 399-408. https://doi.org/10.1080/08880010802107356
PMid:18569842
- Sedky
MS, Rahman HA, Moussa E, et al. Langerhans Cell Histiocytosis (LCH) in
Egyptian Children: Does Reactivation Affect the Outcome? Indian J
Pediatr 2016; 83: 214-9. https://doi.org/10.1007/s12098-015-1801-8
- A
multicentre retrospective survey of Langerhans' cell histiocytosis: 348
cases observed between 1983 and 1993. The French Langerhans' Cell
Histiocytosis Study Group. Arch Dis Child 1996; 75: 17-24. https://doi.org/10.1136/adc.75.1.17
- Aricò
M, Astigarraga I, Braier J, et al; Histiocyte Society. Lack of bone
lesions at diagnosis is associated with inferior outcome in multisystem
langerhans cell histiocytosis of childhood. Br J Haematol 2015;169:
241-8. https://doi.org/10.1111/bjh.13271
PMid:25522229
- Lee
JW, Shin HY, Kang HJ, Kim H, et al. Clinical characteristics and
treatment outcome of Langerhans cell histiocytosis: 22 years'
experience of 154 patients at a single center. Pediatr Hematol Oncol
2014; 31: 293-302. https://doi.org/10.3109/08880018.2013.865095
PMid:24397251
- Postini
AM, del Prever AB, Pagano M, et al. Langerhans cell histiocytosis: 40
years' experience. J Pediatr Hematol Oncol 2012; 34: 353-8. https://doi.org/10.1097/MPH.0b013e318257a6ea
PMid:22627580
- Morren
MA, Vanden Broecke K, Vangeebergen L, et al. Diverse Cutaneous
Presentations of Langerhans Cell Histiocytosis in Children: A
Retrospective Cohort Study. Pediatr Blood Cancer 2016; 63: 486-92. https://doi.org/10.1002/pbc.25834
PMid:26586230
- Badalian-Very
G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans
cell histiocytosis. Blood 2010; 116: 1919-23. https://doi.org/10.1182/blood-2010-04-279083
PMid:20519626 PMCid:PMC3173987
- Berres
ML, Lim KP, Peters T, Price J, et al. BRAF-V600E expression in
precursor versus differentiated dendritic cells defines clinically
distinct LCH risk groups. J Exp Med 2014; 211: 669-83. https://doi.org/10.1084/jem.20130977
PMid:24638167 PMCid:PMC3978272
- Weitzman
S, Braier J, Donadieu J, Egeler RM, Grois N, Ladisch S, Pötschger U,
Webb D, Whitlock J, Arceci RJ. 2'-Chlorodeoxyadenosine (2-CdA) as
salvage therapy for Langerhans cell histiocytosis (LCH). results of the
LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 2009;
53: 1271-6. https://doi.org/10.1002/pbc.22229
PMid:19731321
- Rigaud
C, Barkaoui MA, Thomas C, et al. Langerhans cell histiocytosis:
therapeutic strategy and outcome in a 30-year nationwide cohort of 1478
patients under 18 years of age. Br J Haematol 2016;174: 887-98. https://doi.org/10.1111/bjh.14140
PMid:27273725
- Donadieu
J, Bernard F, van Noesel M, et al. Cladribine and cytarabine in
refractory multisystem Langerhans cell histiocytosis: results of an
international phase 2 study. Blood 2015; 126: 1415-23. https://doi.org/10.1182/blood-2015-03-635151
PMid:26194764 PMCid:PMC4624454
- Rodriguez-Galindo
C, Jeng M, Khuu P, McCarville MB, Jeha S. Clofarabine in refractory
Langerhans cell histiocytosis. Pediatr Blood Cancer 2008; 51: 703-6. https://doi.org/10.1002/pbc.21668
PMid:18623218
- Simko
SJ, Tran HD, Jones J, Bilgi M, Beaupin LK, Coulter D, Garrington T,
McCavit TL, Moore C, Rivera-Ortegón F, Shaffer L, Stork L, Turcotte L,
Welsh EC, Hicks MJ, McClain KL, Allen CE. Clofarabine salvage therapy
in refractory multifocal histiocytic disorders, including Langerhans
cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease.
Pediatr Blood Cancer 2014; 61: 479-87. https://doi.org/10.1002/pbc.24772
PMid:24106153 PMCid:PMC4474604
- Uppuluri
R, Ramachandrakurup S, Subburaj D, Bakane A, Raj R. Excellent remission
rates with limited toxicity in relapsed/refractory Langerhans cell
histiocytosis with pulse dexamethasone and lenalidomide in children.
Pediatr Blood Cancer 2017; 64: 110-2. https://doi.org/10.1002/pbc.26199
PMid:27555565
- Uppuluri
R, Ramachandrakurup S, Balaji R, Subburaj D, Bakane A, Raj R.
Successful Treatment of Refractory Langerhans Cell Histiocytosis of the
Choroid Plexus in a Child With Pulse Dexamethasone and Lenalidomide. J
Pediatr Hematol Oncol 2017; 39: 74-8. https://doi.org/10.1097/MPH.0000000000000735
PMid:28099396
- Heisig
A, Sörensen J, Zimmermann SY, Schöning S, Schwabe D, Kvasnicka,
Raphaela Schwentner HM, Hutter C, and Lehrnbecher T. Vemurafenib in
Langerhans cell histiocytosis: report of a pediatric patient and review
of the literature. Oncotarget 2018; 9: 22236-40. https://doi.org/10.18632/oncotarget.25277
PMid:29774135 PMCid:PMC5955145
- Awada
G, Seremet T, Fostier K, Everaert H, and Neyns B. Long-term disease
control of Langerhans cell histiocytosis using combined BRAF and MEK
inhibition. Blood Adv 2018; 2: 2156-8. https://doi.org/10.1182/bloodadvances.2018021782
PMid:30154124 PMCid:PMC6113614
[TOP]